 , 陈项项, 史亚慧
, 陈项项, 史亚慧 , 刘杨, 刘永德
, 刘杨, 刘永德河南工业大学环境工程学院, 郑州 450001
收稿日期: 2020-12-25; 修回日期: 2021-01-28; 录用日期: 2021-01-28
基金项目: 国家自然科学基金项目(No.51878251);河南省优秀青年自然科学基金项目(No.212300410034);河南省青年自然科学基金项目(No.212300410132)
作者简介: 万东锦(1982-), 女, 教授(博士), E-mail: djwan@haut.edu.cn
通讯作者(责任作者): 史亚慧(1991—), 女, 博士, 讲师, 主要从事新型环境功能材料的研发与应用. E-mail: shiyahui_52@163.com
摘要:采用化学共沉淀法制备磁性钯钴水滑石催化剂(M-Pd/Co@CHT),用于吸附催化还原去除水中的高氯酸盐.同时,研究了不同溶液pH值、催化剂投加量、温度、共存离子和氢气流量等因素的影响,考察了吸附动力学和等温吸附过程.最后对反应前后材料进行表征,以明确吸附催化氢还原反应机理.结果表明:在较宽的pH范围(5~10)内,M-Pd/Co@CHT显示出对高氯酸盐较高、较稳定的吸附效率;伪二阶动力学模型和Langmuir模型能很好地拟合催化剂对高氯酸盐的吸附规律,说明该吸附过程是近似单层的化学吸附,最大单层吸附容量为172.65 mg·g-1;温度、投加量和供氢气量对M-Pd/Co@CHT加氢催化还原高氯酸盐有显著的影响,当高氯酸盐初始浓度为10 mg·L-1时,在最优实验条件下30 min可以去除约54%的高氯酸盐;利用XRD、FTIR、BET、XPS及VSM等手段对M-Pd/Co@CHT进行表征,结果表明,介孔M-Pd/Co@CHT可以有效吸附或催化高氯酸盐;高氯酸盐首先被吸附至M-Pd/Co@CHT上从而恢复其层状结构,而后在Pd/Co二元金属催化剂的作用下被氢还原去除;利用外加磁场能够实现材料的固液分离,可以有效地避免二次污染.
关键词:磁性水滑石Pd/Co吸附氢催化高氯酸盐
Study on the adsorption and catalytic hydrogen reduction of ClO4- from aqueous solution by magnetic hydrotalcite based bimetallic catalyst
WAN Dongjin
, CHEN Xiangxiang, SHI Yahui, LIU Yang, LIU YongdeCollege of Environmental Engineering, Henan University of Technology, Zhengzhou 450001
Received 25 December 2020; received in revised from 28 January 2021; accepted 28 January 2021
Abstract: The magnetic calcined Mg/Fe hydrotalcite-based Pd/Co catalyst (M-Pd/Co@CHT) was synthesized by the co-precipitation method, and was used to remove perchlorate in water through adsorption and catalytic hydrogen reduction. The effect of factors including initial pH, catalyst dosage, temperature, coexisting ions and H2 flow rate on perchlorate removal was investigated, and the adsorption kinetics and isotherm models were fitted. Meanwhile, the characterization of materials before and after reaction was carried out to elucidate the reaction mechanism of adsorption and catalytic hydrogen reduction. The results showed that in the wide pH range (5~10), M-Pd/Co@CHT exhibited a higher adsorption efficiency. Adsorption kinetics and isotherms data satisfactorily followed the pseudo-second-order kinetics model and the Langmuir model, indicating that the adsorption process is a single-layer chemical adsorption process, with maximum adsorption capacity of 176.9 mg·g-1. The temperature, catalyst dosage and H2 flow rate had the significant effect on catalytic reduction removal of perchlorate, and under the optimal experimental conditions, the remove efficiency of perchlorate with initial concentration of 10 mg·L-1 was 54% during 30 min. The results of XRD, FTIR, BET, XPS and VSM indicated that the mesoporous M-Pd/Co@CHT can effectively remove perchlorates by adsorption or catalysis. Specifically, perchlorate was first adsorbed to M-Pd/Co@CHT to restore its layered structure, and then reduced by atomic H at Pd/Co bimetals active sites. The solid-liquid separation of the material can be easily achieved by using an external magnetic field, avoiding secondary pollution effectively.
Keywords: magnetic hydrotalcitePd/Coadsorptionhydrogenation catalysisClO4-
1 引言(Introduction)高氯酸盐(ClO4-)被广泛应用于工业领域, 如固体火箭燃料、烟火及爆炸物的推进剂的生产加工过程中(Xu et al., 2010; Wan et al., 2016; Wan et al., 2018).高氯酸盐在水溶液中具有高溶解性、不挥发性和反应惰性, 容易残留在水环境中造成污染, 一旦被人体摄入可能会通过抑制碘化物的吸收能力而引起甲状腺功能障碍(Charnley, 2008; Pleus et al., 2018; Song et al., 2019), 从而对人体健康产生严重威胁.研究发现(Gan et al., 2014; Kumarathilaka et al., 2016), 高氯酸盐污染遍布世界各地, 在随机抽取的11个国家和地区的水、土壤、空气等环境介质中均检测出了高氯酸盐, 其中, 玻利维亚部分地区的落尘中高氯酸盐含量高达500 mg·kg-1.宇盛好等(2017)对上海市售粮食、水果、蔬菜等8类食品共80件样品的检测结果显示, 高氯酸盐检出率为78.8%, 普通居民的高氯酸盐平均暴露量为每日0.25 μg·kg-1体质量, 高消费人群的暴露量(P95)为每日0.44 μg·kg-1体质量, 已超过欧洲食品安全局设置的高氯酸盐每日容许摄入量(0.3 μg·kg-1体质量), 存在一定的健康风险.
目前, 去除水中高氯酸盐的主要方法包括生物还原、物理法(Baidas et al., 2011)和化学还原法(Parette et al., 2005; Baidas et al., 2011; Ricardo et al., 2012; Xu et al., 2018).其中, 生物法成本低且易于应用(Xu et al., 2016), 但容易受环境因素如温度的影响, 且需要额外的处理工艺进一步对水中微生物进行处理(Yoon et al., 2009).物理法如膜分离和离子交换法仅仅是将高氯酸盐浓缩或转移, 并未实现其形态转化, 且处理过程会产生浓水, 需要进一步处理.化学法由于可实现高氯酸盐的形态转化而受到研究者的关注.化学还原法可以将ClO4-转化为无毒无害的Cl-(Simon et al., 2006; Robert, 2014; Song et al., 2019), 但基于ClO4-具有较高的反应活化能(120 kJ·mol-1), 传统的化学还原过程受动力学限制(Qi et al., 2016).因此, 本研究拟使用催化剂加氢催化还原法来去除水中低浓度的高氯酸盐.
催化加氢还原法是指还原剂提供电子将ClO4-还原为Cl-的过程, 其中常用过渡金属作为直接电子供体或作为催化剂促进电子转移(Gauthard et al., 2003).研究证实, 单金属催化剂(如Pd、Pt)及其双金属体系能够催化加氢有效还原含氧阴离子, 其中, 双金属催化剂的加氢活性显著高于单金属催化剂(Gauthard et al., 2003).Hurley和Shapley (2007)合成的一种非均相双金属催化剂铼/钯活性炭复合材料(Re/Pd-AC)已被用于加氢催化还原高氯酸盐, 该催化剂中Pd组分用于结合并“活化”H2, 使在Pd表面形成H原子, 然后H原子溢出并分散在载体Re-O上与高氯酸盐发生还原反应, 但该催化剂的最佳催化效率(100 mg·g-1)仅在酸性条件下(pH约为3)才能实现.Kim等(2013)和Yao等(2016)合成了吸附/催化双功能材料钯/氮活性炭(Pd/N-AC)用于去除ClO4-, 研究表明, Pd/N-AC中带正电荷的N-官能团为高氯酸盐提供了吸附位点, 吸附饱和后, 在氢气氛围下高氯酸盐被活性炭表面的Pd催化还原为Cl-, 但需要在较高的温度条件下(333 K)才能实现.Mahnudov等(2008)合成了一系列单金属和双金属Pt基催化剂(如Co-Pt/C、W-Pt/C、Pt/C、Ni-Pt/C)用于催化加氢还原高氯酸盐, 研究表明, 相比于其他催化剂(Pt/C、Ni-Pt/C、W-Pt/C), Co-Pt/C具有最高的氢吸附能力和最优的高氯酸盐还原性能, 其中, 第二催化金属Co还原高氯酸盐量占总高氯酸盐还原量的80%~85%, 直接金属Pt还原高氯酸盐量占15%~20%.XPS分析还表明, Pt在单金属和双金属催化剂中以金属形式存在, 而第二金属Co以氧化形式存在(Mahnudov et al., 2008).因此, 本研究选择Pd和Co作为双金属催化剂.与此同时, 载体特性对催化还原效果有着重要的影响.水滑石(阴离子粘土)是一类具有较大的比表面积和较强的阴离子交换能力的无机矿物, 可以作为催化剂载体, 为高氯酸盐的催化加氢还原提供吸附活性位点.
此外, 催化剂的回收和再生问题也是限制其应用的主要因素.基于以上研究背景, 本研究以水滑石为基底合成磁性钯钴水滑石催化剂(M-Pd/Co@HT), 进一步热解处理得到焙烧态催化剂(M-Pd/Co@CHT).以M-Pd/Co@CHT为研究对象, 考察吸附催化氢还原过程对水中高氯酸盐的去除性能, 以期为化学催化法在水体高氯酸盐污染处理中的应用提供理论和实践基础.
2 材料与方法(Materials and methods)2.1 实验材料PdCl2、CoCl2、MgCl2·6H2O、FeCl3·6H2O、NaOH和Na2CO3均为分析纯, ClO4-溶液由NaClO4(优级纯)配制, 纳米Fe3O4(99.5%, 20 nm)购于上海麦克林.实验所需溶液均为去离子水配制.
2.2 材料的制备采用共沉淀法制备M-Pd/Co@HT, 具体操作为:取MgCl2·6H2O (1.2 mol·L-1)、FeCl3·6H2O (0.4 mol·L-1)、PdCl2 (7.5×10-3 mol·L-1)和CoCl2 (1.4×10-2 mol·L-1)配制成溶液A;取足量的NaOH和Na2CO3配制成溶液B;在40 ℃下将溶液A和B迅速滴加到有9.408 g Fe3O4的悬浮液中, 调节pH为10~11, 并剧烈搅拌2 h, 然后置于65 ℃恒温水浴锅中晶化18 h, 所得晶化产物用去离子水反复洗涤至其电导率于2 mS·m-1以下, 置于105 ℃下干燥8 h即得到M-Pd/Co@HT.将M-Pd/Co@HT置于马弗炉中, 在500 ℃条件下煅烧, 冷却后将煅烧产物研磨至粉末状并过100目筛;然后放入管式炉中, 在H2 (50 mL·min-1)和N2 (150 mL·min-1)气氛, 200 ℃条件下还原煅烧产物120 min, 即得到M-Pd/Co@CHT.
2.3 吸附实验M-Pd/Co@CHT对ClO4-的吸附容量按照式(1)进行计算.
 | (1) |
2.3.1 吸附动力学配制10 mg·L-1的ClO4-溶液于500 mL锥形瓶中, 分别加入0.5、1.0、2.0 g·L-1的催化剂, 置于磁力搅拌器上搅拌(25 ℃, 50 r·min-1), 测定0、0.6、1、2、4、6、11和23 h时溶液中ClO4-浓度.
2.3.2 吸附等温线配制一系列不同浓度(50、100、200、300、400、500和600 mg·L-1)的ClO4-溶液(100 mL), 置于250 mL锥形瓶中, 催化剂量投加为1.0 g·L-1, 分别在298、308和318 K下恒温振荡(150 r·min-1) 24 h, 以达到吸附平衡.测定溶液中ClO4-浓度, 平衡吸附量用qe表示.
2.3.3 初始pH值的影响配制10 mg·L-1的ClO4-溶液, 加入0.1 mol·L-1的HCl或NaOH调节pH值.初始pH值范围为3~11, 催化剂投加量为1.0 g·L-1, 在25 ℃下恒温振荡(150 r·min-1) 24 h, 测定溶液中ClO4-浓度及溶液平衡pH值.
2.4 催化实验ClO4-溶液的初始浓度均为10 mg·L-1, 加入到500 mL血清瓶中, 并置于恒温加热磁力搅拌器上控温及搅拌(50 r·min-1), 持续向瓶中通入一定量的氢气进行催化实验.
H2流量的影响:调节H2流量分别为0、10、30、40、50 mL·min-1, 催化剂投加量为1.0 g·L-1, 在25 ℃下恒温搅拌90 min, 测定溶液中ClO4-浓度.
投加量的影响:通入H2流量为50 mL·min-1, 催化剂投加量分别为0、0.5、1.0和2.0 g·L-1, 在25 ℃下恒温搅拌90 min, 测定溶液中ClO4-浓度.
温度的影响:通入H2流量为50 mL·min-1, 催化剂投加量为2.0 g·L-1, 分别在298、308和318 K下恒温搅拌30 min, 测定溶液中ClO4-浓度.
共存离子的影响:分别加入10 mg·L-1的Cl-、NO3-、SO42-和PO43-溶液, 催化剂投加量为1.0 g·L-1, 在25 ℃下恒温搅拌90 min, 测定溶液中ClO4-浓度.
2.5 分析与表征ClO4-水样经0.22 μm滤膜过滤后采用离子色谱仪(ICS-2100, 美国Dionex)测定其浓度.采用X射线衍射仪(XRD-6100, 日本岛津)进行XRD测试, X射线源为Cu靶Kα射线, 管压40 kV, 管流30 mA, 扫描步长0.02°, 扫描速度为10°·min-1.FTIR测量采用傅里叶变换红外光谱仪(Prestige-21, 日本岛津), 材料经KBr压片处理, 扫描范围为500~4000 cm-1.采用全自动比表面积及孔隙度分析仪(ASAP2020 HD88, 美国麦克)测定材料的比表面积和孔径.采用电感耦合等离子体光谱仪(安捷伦5100, ICP-OES)测定溶液中的钴元素浓度.
2.6 M-Pd/Co-CHT的重复利用性能采用0.1 mol·L-1 NaOH溶液作为再生液.将吸附-还原后的M-Pd/Co-CHT加入到NaOH溶液中, 在25 ℃恒温水浴振荡器中振荡24 h进行再生.在25 ℃下测试再生的M-Pd/Co-CHT对10 mg·L-1 ClO4-的去除性能(催化剂投加量为2.0 g·L-1, 氢气流量为50 mL·min-1, 反应时间90 min).
3 结果与讨论(Results and discussion)3.1 吸附实验3.1.1 吸附动力学25 ℃条件下, M-Pd/Co@CHT对水溶液中ClO4-的吸附容量随时间的变化情况如图 1所示.为了对动力学过程进行定量描述, 采用一阶动力学(式(1))、假二阶动力学(式(2))和粒内扩散模型(式(3))对实验结果进行拟合.
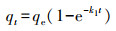 | (1) |
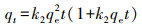 | (2) |
 | (3) |
 |
| 图 1 M-Pd/Co@CHT对ClO4-的吸附动力学过程及其拟合 Fig. 1Adsorption kinetics of ClO4- onto M-Pd/Co@CHT |
式中, qe和qt分别为吸附平衡时和t时刻的吸附容量(mg·g-1), t为反应时间(min), k1 (min-1)、k2 (g·mg-1·min-1)和k3 (mg·g-1·min-0.5)分别为一阶动力学、假二阶动力学及粒内扩散模型的吸附速率常数.初始吸附速率由如下公式进行计算:
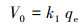 | (4) |
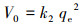 | (5) |
表 1(Table 1)
| 表 1 动力学模型拟合参数 Table 1 Experimental data and the fitting parameters of three kinetics models | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 1 动力学模型拟合参数 Table 1 Experimental data and the fitting parameters of three kinetics models
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1.2 吸附等温线在298、308和318 K条件下, M-Pd/Co@CHT对ClO4-的吸附等温线如图 2所示.采用Langmuir模型(式(6))和Freundlich模型(式(7))对实验数据进行拟合, 拟合参数结果如表 2所示.Langmuir模型可用于描述吸附剂表面均一的单分子层吸附, Freundlich模型用于描述在非均质表面发生的吸附.
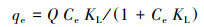 | (6) |
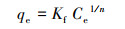 | (7) |
 |
| 图 2 M-Pd/Co@CHT对ClO4-的吸附等温线拟合结果 Fig. 2Adsorption isotherm of ClO4- onto M-Pd/Co@CHT |
表 2(Table 2)
| 表 2 等温线模型拟合参数 Table 2 Adsorption isotherm parameters for M-Pd/Co@CHT | ||||||||||||||||||||||||||||||||||||
表 2 等温线模型拟合参数 Table 2 Adsorption isotherm parameters for M-Pd/Co@CHT
| ||||||||||||||||||||||||||||||||||||
式中, qe为吸附平衡时的吸附容量(mg·g-1), Q为吸附剂的单层吸附容量(mg·g-1), Ce为ClO4-的吸附平衡浓度(mg·L-1), KL为Langmuir常数(L·mg-1), Kf(mg·g-1)(L·mg-1)n和n为Freundlich相关常数.
结果表明, 随着温度从298 K升高到318 K, 最大单层吸附容量Q从152.79 mg·g-1上升到172.65 mg·g-1, 表明升温可以增强M-Pd/Co@CHT的吸附性能.由表 2可知, 与Freundlich模型相比, Langmuir模型更适合用来描述M-Pd/Co@CHT的吸附等温线过程(R2>0.99), 说明M-Pd/Co@CHT对ClO4-的吸附符合单层吸附假设.
为了进一步探讨M-Pd/Co@CHT对ClO4-的吸附热力学过程, 引入吉布斯自由能(ΔG0), 如式(8)所示.
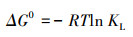 | (8) |
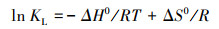 | (9) |
表 3(Table 3)
| 表 3 M-Pd/Co@CHT对ClO4-的吸附热力学参数 Table 3 Thermodynamic parameters for ClO4- adsorbed by M-Pd/Co@CHT | ||||||||||||||||
表 3 M-Pd/Co@CHT对ClO4-的吸附热力学参数 Table 3 Thermodynamic parameters for ClO4- adsorbed by M-Pd/Co@CHT
| ||||||||||||||||
3.1.3 初始pH的影响从图 3可以看出, M-Pd/Co@CHT的吸附容量受溶液初始pH值的影响, 随着溶液初始pH值的上升, 吸附容量呈现先上升后下降的趋势.在酸性较大条件下, 材料可能被破坏失去吸附能力(顾颐冰等, 2016).pH在5~10时, M-Pd/Co@CHT对ClO4-具有稳定的吸附容量(5.60~6.54 mg·g-1), 表明在较宽的pH范围M-Pd/Co@CHT具有较好的ClO4-吸附性能.因此, 整个实验过程中没有调节溶液初始pH, 避免引入其他离子影响实验结果.
图 3(Fig. 3)
 |
| 图 3 溶液初始pH对ClO4-吸附容量的影响 Fig. 3Effect of initial pH on the adsorption of ClO4- at 25 ℃ |
3.2 催化性能测试3.2.1 H2流量的影响研究H2流量对催化氢还原去除ClO4-的影响, 结果如图 4a所示.在未通氢气的情况下, ClO4-仅通过M-Pd/Co@CHT的吸附作用去除, 而在通入H2后, ClO4-将通过吸附催化氢还原双重作用得以去除.在25 ℃, 投加量为1 g·L-1条件下, 至90 min时, ClO4-的吸附去除率仅为12.09%.在相同条件下, 当通入H2流量为10 mL·min-1时, 反应90 min时ClO4-的去除率为31.63%, 是吸附去除率的2.62倍.由图 4a可知, 随着通入H2流量的增大, ClO4-的去除率有所提高.当H2流量为50 mL·min-1时, 反应90 min时ClO4-的去除率最大, 为41.22%.H2流量增大, 还原H的供给更为充分, 从而导致ClO4-去除率升高.
图 4(Fig. 4)
 |
| 图 4 H2流量(a)、M-Pd/Co@CHT投加量(b)、温度(c)及共存离子(d)对去除ClO4-的影响 Fig. 4Effect of H2 flow rate(a), catalyst dosage(b), temperature(c) and co-existing ions(d) on the removal of ClO4- by M-Pd/Co@CHT |
3.2.2 投加量的影响研究不同的M-Pd/Co@CHT投加量对去除ClO4-的影响, 结果如图 4b所示.当未加入M-Pd/Co@CHT时, 仅通入50 mL·min-1的H2, ClO4-几乎不被去除.当M-Pd/Co@CHT的投加量从0.5 g·L-1增加到2.0 g·L-1时, 在90 min内ClO4-去除效率从21.11%增加到56.60%.此外, 在反应初始60 min内, ClO4-去除速率较快, 而后ClO4-的去除趋势趋于平缓.
3.2.3 温度的影响研究温度对催化剂去除ClO4-的影响, 结果如图 4c所示.随着水温从25 ℃升高至45 ℃, 反应30 min, ClO4-去除率从37.13%提高到53.70%.当温度为45 ℃时, 反应20 min, ClO4-去除率增大到48.30%, 随后10 min, ClO4-的去除逐渐趋于平缓, 最终去除率为53.70%.结合吸附等温过程可知, 高氯酸盐的吸附和催化还原均为吸热过程, 温度的升高利于高氯酸盐的吸附和加氢催化还原反应.
3.2.4 共存离子的影响高氯酸盐污染水体中普遍存在着大量的阴离子, 它们的存在可能影响高氯酸盐的还原.因此, 选取水体中常见的Cl-、NO3-、SO42-和PO43-作为共存离子, 考察其对高氯酸盐吸附催化氢还原的影响, 结果如图 4d所示.当高氯酸盐与Cl-、NO3-、SO42-和PO43-共存时, 其去除率明显降低.反应90 min, 高氯酸盐去除率从41.21%分别降低到33.31%、23.22%、17.56%和12.49%.共存阴离子对高氯酸盐还原的影响顺序为:PO43->SO42- >NO3->Cl-.有研究表明, 水滑石类化合物对阴离子的吸附主要受静电引力的影响, 阴离子价态越高, 占据的吸附点位越多, 对吸附的影响也就越大(许云峰等, 2014).本研究中, 高价态的阴离子对催化还原效果影响较大, 其原因与吸附过程类似, 高价态阴离子占据较多吸附点位, 进而影响高氯酸盐在水滑石表面的进一步催化还原.
3.2.5 M-Pd/Co@CHT的重复利用性能可重复使用性能是评估材料经济适用性的重要参数, 使用NaOH溶液(0.1 mol·L-1)对吸附-还原后的M-Pd/Co@CHT进行再生, 再生后的M-Pd/Co@CHT对ClO4-的去除性能结果如图 5所示.经过3次循环使用后, M-Pd/Co@CHT对ClO4-的去除率由57.1%下降到9.8%.这是由于催化氢还原ClO4-后的材料(M-Pd/Co@CHT-R)恢复层状结构(XRD已证实), Pb/Co活性位点被ClO4-、Cl-占据, 导致催化性能下降;同时, 有研究证实(Yang et al., 2017), 随NaOH溶液浓度的增加, 材料表面附着离子的脱附效率提高, 再生水滑石的催化性能增强, 而本研究的材料再生过程中, 选用的NaOH溶液浓度相对较低(0.1 mol·L-1), 导致M-Pd/Co@CHT的再生效果不佳.同时, M-Pd/Co@CHT催化氢还原反应后溶液中Co浓度低于检测限值(0.02 mg·L-1).因此, M-Pd/Co@CHT是一种环保型吸附-还原催化材料.
图 5(Fig. 5)
 |
| 图 5 M-Pd/Co@CHT的重复利用性能 Fig. 5Reuse performance of M-Pd/Co@CHT on the reduction of perchlorate |
4 材料表征及反应机理分析(Material characterization and reaction mechanism analysis)4.1 XPS分析使用XPS分析材料M-Pd/Co@CHT中各元素状态和含量及Pd、Co和Fe的结合能.如图 6a所示, Mg 2p、C 1s、Pd 3d、O 1s、Fe 2p和Co 2p是主要峰, 表明Pd和Co已成功置入到催化剂中(Liu et al., 2020).在图 6b中, Fe 2p被很好地拟合成6个峰, 其中, 710.3 eV和723.8 eV处的峰是Fe(Ⅱ)的特征峰, 711.7 eV和725.2 eV处的峰是Fe(Ⅲ)的特征峰, 而717.8 eV处的峰是Fe(Ⅱ)和Fe(Ⅲ)共同的卫星峰.在图 6c中, Pd 3d被很好地拟合成两个峰, 其中, 335.51 eV处的峰是Pd(0)的特征峰, 约占3/4, 337.50 eV处的弱峰是Pd(Ⅱ)的特征峰, 仅约占1/4, 这表明制备过程中钯化合物被H2充分还原为零价Pd.据报道, 778.40 eV对应Co(0)的特征峰(Mahnudov et al., 2008).由图 6d可知, 未观察到Co(0)的特征峰, 结合能为782.3 eV和785.1 eV的峰分别对应Co(Ⅱ)2p3/2和Co(Ⅲ)2p3/2, 结合能为787 eV处的峰为Co(Ⅱ)和Co(Ⅲ)共同的卫星峰(Lan et al., 2014; Liu et al., 2020), 因此, 本材料中第二种贵金属(Co)的化合价态均为氧化态, 用作保护金属.
图 6(Fig. 6)
 |
| 图 6 M-Pd/Co@CHT XPS总谱(a)、Fe2p分谱(b)、Pd3d分谱(c)及Co2p分谱(d) Fig. 6XPS survey(a), Fe2p (b), Pd3d(c) and Co2p(d) of M-Pd/Co@CHT |
4.2 XRD分析M-Pd/Co@HT的XRD图谱如图 7所示, 根据标准卡PDF#46-0203, 在2θ为12.31°、24.98°和59.18°处分别为(003)、(006)和(213)晶面的衍射峰, 代表典型水滑石对称结构;根据标准卡PDF#88-2182和PDF#70-2674, 在2θ为43.07°和57.42°处分别为(400)和(018)衍射峰, 代表混合氧化物(Mg(Fe)O).将材料煅烧后, M-Pd/Co@HT的(003)、(006)和(213)晶面的衍射峰被明显减弱, 这表明水滑石的典型层状结构被破坏.根据标准卡PDF#99-0073, 在2θ为30.08°(220)、35.43°(311)和62.52°(440)处的衍射峰是Fe3O4的典型特征峰, 表明Fe3O4被成功负载到材料中, 使材料具备磁性(Liu et al., 2020).值得注意的是, 在XRD图谱中, 没有出现相关Pd/Co的特征峰, 可能是因为Pd/Co的含量较低且分散性良好.吸附高氯酸盐后, 观测到M-Pd/Co@CHT-A的(003)、(006)和(213)衍射峰, 表明吸附过程伴随着水滑石层状结构的恢复, 水中的高氯酸盐被吸附在煅烧后的水滑石表面并形成新的阴离子层(Maity et al., 2007).还原高氯酸盐后的材料(M-Pd/Co@CHT-R)与吸附后的材料(M-Pd/Co@CHT-A)峰形类似, 表明催化还原过程也伴随着层状结构的重建.根据M-Pd/Co@CHT-R的图谱, 在2θ=20°处出现一个新的峰, 推测是Mg2(OH)3Cl.在催化还原过程中, ClO4-首先被吸附到M-Pd/Co@CHT材料上使其恢复层状结构;而后ClO4-在Pd/Co二元金属催化剂的作用下被氢还原为Cl-, 形成Mg2(OH)3Cl.
图 7(Fig. 7)
 |
| 图 7 焙烧前后(M-Pd/Co@HT和M-Pd/Co@CHT)及分别吸附和还原后的材料(M-Pd/Co@CHT-A和M-Pd/Co@CHT-R)的XRD光谱 Fig. 7XRD spectra of the adsorbed and reduced materials (M-Pd/Co@CHT-A and M-Pd/Co@CHT-R) before and after roasting (M-Pd/Co@HT, M-Pd/Co@CHT) respectively |
4.3 FTIR分析材料的红外FTIR光谱如图 8所示.对于M-Pd/Co@HT, 在3471、1643和1371 cm-1处有3个峰, 分别对应于—OH、层间的H2O和CO32-的拉伸振动;煅烧后M-Pd/Co@CHT的—OH和CO32-峰减弱, 表明在煅烧过程中M-Pd/Co@HT失去了部分层间水和CO32-(陈慧琴等, 2006).吸附高氯酸盐后, M-Pd/Co@CHT-A在1093 cm-1处出现ClO4-的特征峰, 表明ClO4-被吸附到材料上.经加氢还原后, 材料M-Pd/Co@CHT-R中1093 cm-1处ClO4-的峰值减弱, 表明吸附到材料上的ClO4-被还原为Cl-.综合以上分析, M-Pd/Co@CHT加氢催化还原去除高氯酸盐包括两个主要步骤:第一, 高氯酸盐被吸附至M-Pd/Co@CHT表面形成阴离子层, 从而重建水滑石的层状结构;第二, 吸附的高氯酸盐在催化剂表面的Pd/Co活性位点上被氢还原为Cl-.
图 8(Fig. 8)
 |
| 图 8 焙烧前后(M-Pd/Co@HT和M-Pd/Co@CHT)及分别吸附和还原后材料(M-Pd/Co@CHT-A和M-Pd/Co@CHT-R)的FTIR光谱 Fig. 8FTIR spectra of the adsorbed and reduced materials (M-Pd/Co@CHT-A and M-Pd/Co@CHT-R) before and after roasting (M-Pd/Co@HT, M-Pd/Co@CHT) respectively |
4.4 BET分析M-Pd/Co@CHT对N2的吸附脱附等温线如图 9所示.BET分析结果表明, M-Pd/Co@CHT是介孔材料(王颖等, 2006) (平均孔径40.63 nm, 比表面积75.36 m2·g-1, 孔体积0.35 cm3·g-1), 可以为吸附高氯酸盐提供充足的吸附位点.
图 9(Fig. 9)
 |
| 图 9 M-Pd/Co@CHT的N2吸附-脱附等温线 Fig. 9N2 adsorption-desorption isotherm of M-Pd/Co@CHT |
4.5 VSM分析M-Pd/Co@CHT的磁滞回线如图 10所示, 可以看出, 达到饱和状态的磁化强度为10.94 emu·g-1.M-Pd/Co@CHT的矫顽力接近于零, 并且磁化强度随外部磁场的增加而增大.因此, 在外加磁场的作用下, M-Pd/Co@CHT可以聚集在磁铁周围, 即在吸附或催化高氯酸盐之后利用外加磁场能够实现材料的固液分离(廖琴瑶等, 2019; 高华敏等, 2020).
图 10(Fig. 10)
 |
| 图 10 M-Pd/Co@CHT的磁滞回线图 Fig. 10Hysteresis loop diagrams for M-Pd/Co@CHT |
5 结论(Conclusions)1) 吸附实验结果表明, 在投加量为1 g·L-1, 反应温度为45 ℃条件下, M-Pd/Co@CHT对高氯酸盐的最大吸附容量为172.65 mg·g-1, 在较宽的pH范围(5~10)内, M-Pd/Co@CHT对高氯酸盐有稳定的吸附效率.
2) 伪二阶动力学模型和Langmuir模型能够很好地拟合M-Pd/Co@CHT对高氯酸盐的吸附规律, 说明该吸附过程是近似单层的化学吸附.
3) 加氢催化实验结果表明, 温度、投加量和氢气供给量对M-Pd/Co@CHT加氢催化去除高氯酸盐有显著的影响, 提高温度、增加催化剂投加量和氢气供给量可以促进M-Pd/Co@CHT对高氯酸盐的去除.
4) XRD、FTIR、BET、XPS、VSM分析结果表明, M-Pd/Co@CHT加氢催化还原去除高氯酸盐包括吸附和催化两个主要步骤;M-Pd/Co@CHT具有介孔结构(40.63 nm), 可以有效吸附或催化高氯酸盐, 在去除高氯酸盐后利用外加磁场能够实现材料的固液分离.因此, M-Pd/Co@CHT可以作为一种有效的新型材料去除水中的高氯酸盐.
参考文献
| Baidas S, Gao B, Meng X. 2011. Perchlorate removal by quaternary amine modified reed[J]. Journal of Hazardous Materials, 189(1): 54-61. |
| 陈慧琴, 詹正坤. 2006. 钴镁铁水滑石的合成, 表征及衍生复合氧化物酸碱催化活性研究[J]. 化学研究与应用, (10): 1189-1192. DOI:10.3969/j.issn.1004-1656.2006.10.011 |
| Charnley G. 2008. Perchlorate: Overview of risks and regulation[J]. Food & Chemical Toxicology, 46(7): 2307-2315. |
| Gan Z, Sun H, Wang R, et al. 2014. Occurrence and exposure evaluation of perchlorate in outdoor dust and soil in mainland China[J]. The Science of the Total Environment, 470-471: 99-106. DOI:10.1016/j.scitotenv.2013.09.067 |
| Gauthard F, Epron F, Barbier J. 2003. Palladium and platinum-based catalysts in the catalytic reduction of nitrate in water: effect of copper, silver, or gold addition[J]. Journal of Catalysis, 220(1): 182-191. DOI:10.1016/S0021-9517(03)00252-5 |
| 高华敏, 石英辉, 李宏林. 2020. 均匀共沉淀法合成磁性水滑石的结构, 性能及应用[J]. 中国陶瓷, 56(8): 45-51. |
| 顾怡冰, 马邕文, 万金泉, 等. 2016. 类水滑石复合材料吸附去除水中硫酸根离子[J]. 环境科学, 37(3): 1000-1007. |
| Hurley K D, Shapley J R. 2007. Efficient heterogeneous catalytic reduction of perchlorate in water[J]. Environmental Science & Technology, 41(6): 2044-2049. |
| Kim Y N, Lee Y, Choi M. 2013. Complete degradation of perchlorate using Pd/N-doped activated carbon with adsorption/catalysis bifunctional roles[J]. Carbon, 65(12): 315-323. |
| Kumarathilaka P, Oze C, Indraratne S P, et al. 2016. Perchlorate as an emerging contaminant in soil, water and food[J]. Chemosphere, 150: 667-677. DOI:10.1016/j.chemosphere.2016.01.109 |
| Lan D, Chen Y, Chen P, et al. 2014. Mesoporous CoO nanocubes@continuous 3D porous carbon skeleton of rose-based electrode for high-performance supercapacitor[J]. ACS Applied Materials & Interfaces, 6(15): 11839-11845. |
| Liu H, Huang Z, Guo B. 2014. Cu/Co/Cr nanocomposites obtained from hydrotalcite precursors as catalysts for thermal decomposition of ammonium perchlorate[J]. Acta Metallurgica Sinica: English letters, 27(2): 211-216. DOI:10.1007/s40195-014-0036-4 |
| Liu Y, Li J, Wu L, et al. 2020. Magnetic spent bleaching earth carbon (Mag-SBE@C) for efficient adsorption of tetracycline hydrochloride: Response surface methodology for optimization and mechanism of action[J]. The Science of the Total Environment, 722: 137817. DOI:10.1016/j.scitotenv.2020.137817 |
| Liu Y, Liu Y, He Q, et al. 2020. Utilizing the "memory effect" of bimetallic-supported hydrotalcites for adsorption and reduction of perchlorate in water[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 593: 124641. DOI:10.1016/j.colsurfa.2020.124641 |
| 廖琴瑶, 袁东, 李佳祁, 等. 2019. 磁性镍铬水滑石的制备及性能研究[J]. 现代化工, 39(11): 127-131. |
| Mahmudov R, Shu Y, Rykov S, et al. 2008. The reduction of perchlorate by hydrogenation catalysts[J]. Applied Catalysis.B, Environmental, 81(1): 78-87. |
| Maity D, Agrawal D C. 2007. Synthesis of iron oxide nanoparticles under oxidizing environment and their stabilization in aqueous and non-aqueous media[J]. Journal of Magnetism and Magnetic Materials, 308(1): 46-55. DOI:10.1016/j.jmmm.2006.05.001 |
| Parette R, Cannon F S, Weeks K. 2005. Removing low ppb level perchlorate, RDX, and HMX from groundwater with cetyltrimethylammonium chloride (CTAC) pre-loaded activated carbon[J]. Water Research, 39(19): 4683-4692. DOI:10.1016/j.watres.2005.09.014 |
| Pleus R C, Corey L M. 2018. Environmental exposure to perchlorate: A review of toxicology and human health[J]. Toxicology and Applied Pharmacology, 358: 102-109. DOI:10.1016/j.taap.2018.09.001 |
| Qi Y, Yao F, Yu Z, et al. 2016. Catalytic and electrocatalytic reduction of perchlorate in water-A review[J]. Chemical Engineering Journal, 306: 1081-1091. DOI:10.1016/j.cej.2016.08.041 |
| Ricardo A R, Carvalho G, Velizarov S, et al. 2012. Kinetics of nitrate and perchlorate removal and biofilm stratification in an ion exchange membrane bioreactor[J]. Water Research, 46(14): 4556-4568. DOI:10.1016/j.watres.2012.05.045 |
| Robert D A J T. 2014. Process control factors for continuous microbial perchlorate reduction in the presence of zero-valent iron[J]. Frontiers of Environmental Science & Engineering, 8(3): 386-393. |
| Simon R, Weber E J. 2006. Reduction of perchlorate in river sediment[J]. Environmental Toxicology and Chemistry, 25(4): 899-903. DOI:10.1897/05-090R.1 |
| Song S, Ruan J, Bai X, et al. 2019. One-step sample processing method for the determination of perchlorate in human urine, whole blood and breast milk using liquid chromatography tandem mass spectrometry[J]. Ecotoxicology and Environmental Safety, 174: 175-180. DOI:10.1016/j.ecoenv.2019.02.081 |
| Song W, Gao B, Zhang X, et al. 2019. Biological reduction of perchlorate in domesticated activated sludge considering interaction effects of temperature, pH, electron donors and acceptors[J]. Process Safety and Environmental Protection, 123: 169-178. DOI:10.1016/j.psep.2019.01.009 |
| Wan D, Liu Y, Wang Y, et al. 2016. Simultaneous bio-autotrophic reduction of perchlorate and nitrate in a sulfur packed bed reactor: Kinetics and bacterial community structure[J]. Water Research, 108: 280-292. |
| Wan D, Wu L, Liu Y, et al. 2018. Adsorption of low concentration perchlorate from aqueous solution onto modified cow dung biochar: Effective utilization of cow dung, an agricultural waste[J]. Science of the Total Environment, 636: 1396. DOI:10.1016/j.scitotenv.2018.04.431 |
| 王颖, 曲久辉, 刘会娟, 等. 2006. Pd-Cu/水滑石吸附催化氢还原水中的硝酸根[J]. 科学通报, (7): 786-791. |
| Xu J, Gao N, Tang Y, et al. 2010. Perchlorate removal using granular activated carbon supported iron compounds: Synthesis, characterization and reactivity[J]. Journal of Environmental Sciences, 22(11): 1807-1813. |
| Xu J, Gao N, Zhao D, et al. 2018. Bromate reduction and reaction-enhanced perchlorate adsorption by FeCl3-impregnated granular activated carbon[J]. Water Research, 149: 149-158. |
| Xu J H, Gao N Y, Zhao D Y, et al. 2016. Different iron salt impregnated granular activated carbon (Fe-GAC) for perchlorate removal: Characterization, performance and mechanism[J]. Colloids & Surfaces A Physicochemical & Engineering Aspects, 509: 99-108. |
| 许云峰, 兰微, 佟天宇, 等. 2014. 水滑石类材料及其对水中阴离子污染物的吸附[J]. 广州化工, 42(21): 22-23. |
| Yang Y, Wen D, Ding Q, et al. 2017. Adsorption behavior of perchlorate removal from aqueous solution using MgAlCe hydrotalcite-like compounds[J]. Desalination and Water Treatment, 88: 257-267. |
| Yao F, Zhong Y, Yang Q, et al. 2016. Effective adsorption/electrocatalytic degradation of perchlorate using Pd/Pt supported on N-doped activated carbon fiber cathode[J]. Journal of Hazardous Materials, 323(B): 602-610. |
| Yoon I H, Meng X, Chao W, et al. 2009. Perchlorate adsorption and desorption on activated carbon and anion exchange resin[J]. Journal of Hazardous Materials, 164(1): 87-94. |
| 宇盛好, 李亦奇, 张旭晟, 等. 2017. 上海市市售食品中高氯酸盐污染的暴露评估[J]. 上海预防医学, 29(6): 426-430. |
