 , 谢莹莹2, 叶翰1, 张金莲1, 易筱筠1, 党志1, 卢桂宁1
, 谢莹莹2, 叶翰1, 张金莲1, 易筱筠1, 党志1, 卢桂宁1
1. 华南理工大学环境与能源学院, 工业聚集区污染控制与生态修复教育部重点实验室, 广州 510006;
2. 韩山师范学院化学与环境工程学院, 潮州 521041
收稿日期: 2020-01-07; 修回日期: 2020-02-14; 录用日期: 2020-02-14
基金项目: 国家自然科学基金(No.41720104004,41931288);广东省重点领域研发项目(No.2019B110207001)
作者简介: 唐苑君(1994-), 女, E-mail:201720141848@mail.scut.edu.cn
通讯作者(责任作者): 卢桂宁, E-mail:lutao@scut.edu.cn
摘要:黄钾铁矾能够通过吸附和共沉淀作用固定酸性矿山废水(AMD)中的重(类)金属(如砷),降低其迁移性和生物可利用性.草酸盐广泛存在于天然水环境中,其具有的羧酸官能团能改变铁矿物的稳定性,进而影响吸持的重(类)金属的再分配行为.利用水热法合成含As(V)黄钾铁矾,探究其在不同草酸盐浓度与pH条件下的溶解、重结晶和共沉淀As(V)的行为.研究结果表明,草酸盐与含As(V)黄钾铁矾表面Fe(III)活性位点配位形成的可溶性强络合物是促进矿物溶解的第一步和关键;在pH 2.5时,含As(V)黄钾铁矾的溶解速率随草酸盐浓度增加而增加,伴随大量As(V)释放到溶液,反应过程中只有少量As(V)重新吸附到固相上,这是由于草酸盐与As(V)竞争矿物表面的同一活性位点;在pH 6.5条件下,草酸盐促进含As(V)黄钾铁矾的重结晶,经X射线衍射分析表明针铁矿和纤铁矿为主要产物,能有效地吸附释放的As(V).研究结果有助于揭示在AMD环境下黄钾铁矾沉积物与含羧酸官能团有机酸共存时对As(V)的释放和固定机理,对AMD环境中As(V)污染控制有重要意义.
关键词:黄钾铁矾草酸盐砷溶解次生矿物
Phase transformation process of As(V)-bearing jarosite and redistribution behavior of As(V) under the oxalate mediating
TANG Yuanjun1
, XIE Yingying2, YE Han1, ZHANG Jinlian1, YI Xiaoyun1, DANG Zhi1, LU Guining11. The Key Laboratory of Pollution Control and Ecosystem Restoration in Industrial Clusters, Ministry of Education, School of Environment and Energy, South China University of Technology, Guangzhou 510006;
2. School of Chemistry and Environmental Engineering, Hanshan Normal University, Chaozhou 521041
Received 7 January 2020; received in revised from 14 February 2020; accepted 14 February 2020
Abstract: Jarosite is considered as an effective scavenger for arsenic (As) under acid mine drainage (AMD) environment due to its strong adsorption capacity and ability to incorporate metal(loid)s. Oxalate is commonly found in natural water and exhibits a prominent contribution to the dissolution of iron minerals. In this study, the influences of oxalate on the dissolution and phase transformation of As(V)-bearing jarosite, as well as redistribution behavior of coprecipitated As(V) were investigated at pH 2.5 and 6.5 under aerobic condition. Results showed that the formation of Fe(III)-oxalate complex promoted the dissolution of As(V)-bearing jarosite and the release of As(V) under pH 2.5 condition. The insignificant re-adsorption of arsenate could be explained by the competition for Fe(III) active sites on the jarosite surface with oxalate. In addition, X-ray diffraction analysis result indicated that As(V)-bearing jarosite transformed to goethite and lepidocrocite at pH 6.5 and oxalate enhanced the crystallization of secondary minerals. After 60 d of reaction, almost all of the liberated As(V) re-adsorbed to the solid phase. It indicates that newly formed goethite and lepidocrocite play a vital role in controlling the fate of As(V). The reaction process involves that ligand promoted dissolution and recrystallization of As(V)-bearing jarosite, while As(V) is effectively immobilized by goethite and lepidocrocite. The present study reveals that carboxylic group in dissolved organic acid can exert great influence on the stability of jarosite and partitioning behavior of coprecipitated As(V) under AMD environment, which is of great significance for arsenic pollution control.
Keywords: jarositeoxalatearsenicdissolutionsecondary minerals
1 引言(Introduction)黄钾铁矾(Jarosite)形成于富铁高硫酸根的酸性环境中, 如酸性矿山废水(AMD, acid mine drainage)和酸性硫酸盐土壤(AAS, acid sulfate soil), 是一种亚稳定的铁羟基硫酸盐矿物, 典型化学式为KFe3(SO4)2(OH)6(Baron et al., 1996; 刘奋武等, 2015).黄钾铁矾在形成过程中, 结构中的K+、Fe3+和SO42-能够分别被与其离子半径和所带电荷相似的Ag+和Pb2+、Al3+和Cu2+、AsO43-和CrO42-等离子部分置换进入到矿物结构中(Forray et al., 2014); 同时黄钾铁矾具有较大的比表面积和丰富的≡Fe—OH官能团(Yang et al., 2015; Gao et al., 2019), 因此, 黄钾铁矾可通过共沉淀或表面吸附的方式固定重金属, 从而使其演变为重(类)金属的沉积库, 如砷、锑、锌、钒等(Welch et al., 2007; Forray et al., 2014; Karimian et al., 2017).
砷(As)是常见的环境毒物和人类致癌物, 广泛分布于矿区河流与农田环境, 所以砷在水体和沉积物中的迁移备受关注(Ren et al., 2018). As的生物可利用性及环境风险与其存在形式和价态紧密相关, As(III)毒性和迁移性比As(V)强; 当环境中氧化还原电位较高时, As在酸性和弱酸性条件下(pH < 6.9)主要以H2AsO4-的形态存在(Fan et al., 2019b).研究表明, 在AMD环境中As污染主要存在于河流沉积物中, 并且主要以残渣态的形态存在(Xie et al., 2018). Savage等(2000)调查发现在加利福利亚南部矿区, 富含黄钾铁矾的沉积物中As含量高达2000 ppm.黄钾铁矾能富集、钝化水体中的As, 对矿区环境中砷污染表现出显著的修复能力.但随着环境条件的改变, 如pH和微生物群落的变化、溶解性有机酸浓度和种类的改变等, 均会使黄钾铁矾发生溶解和相转变, 并影响吸持的砷的迁移行为和毒性(Wang et al., 2017; Gao et al., 2019; Fang et al., 2019).
溶解性有机酸源于根系分泌物和微生物分解的腐殖质, 富含羟基、羧基等活性官能团(Strobel, 2001), 对铁矿物有较高的亲和力, 可改变其稳定性, 从而对Fe的生物地球化学循环过程和重(类)金属迁移产生重大影响(Yu et al., 2013; Xie et al., 2017).草酸普遍存在于自然水环境中(2.5×10-5~4.0×10-3 mol·L-1), 能与重金属形成稳定络合物(Reichard et al., 2007).有研究报道, 在弱酸性有氧条件下, 草酸促进针铁矿溶解, 但抑制针铁矿中共沉淀Se(IV)的释放, 这是由于草酸促进表面可交换态Se(IV)向结合态(≡Fe(SeO)O—CO)和残渣态转化(Fang et al., 2019); 在pH 3条件下, 吸附了As(V)的施氏矿物在草酸盐(1 mmol·L-1)介导下, 反应过程中溶液中总铁含量不断增加, 并且主要以Fe(III)形式存在, 同时促进As(V)从矿物相迁移至溶液相(Ren et al., 2018); Yu等(2013)的研究表明, 在pH 3.0、7.5和11.0条件下, 草酸盐抑制释放到溶液中的As(V)重新吸附到赤铁矿上.草酸在不同体系下促进针铁矿、施氏矿物、赤铁矿的溶解通常是一个表面控制过程(Paris et al., 2013); 同时草酸能与重金属离子、含氧阴离子竞争矿物表面的反应活性位点(Gadol et al., 2017), 竞争吸附反应可以抑制或促进铁矿物溶解, 取决于形成的络合物类型和键的强度(Eick et al., 1999).
目前还没有关于含As(V)黄钾铁矾在草酸盐介导下的地球化学行为研究, 黄钾铁矾转化产物以及As(V)价态变化和迁移情况仍是不清楚的.本文主要是基于矿区酸性河流环境中, 黄钾铁矾沉积物、As(V)和草酸盐共存的情况下, 研究含As(V)黄钾铁矾在不同草酸盐浓度体系下的稳定性, 以及共沉淀砷的分配过程和价态变化, 有助于揭示含羧酸官能团有机酸对负载As(V)黄钾铁矾的影响机制.
2 材料与方法(Materials and methods)2.1 实验材料实验用七水砷酸氢二钠(Na2HAsO4·7H2O, >99%), 2-(N-吗啡啉)乙磺酸(MES, C6H13NO4S·H2O, ≥99%)和醋酸铵(C2H7NO2, ≥98%)均购于阿拉丁试剂有限公司; 草酸钠(Na2C2O4, ≥99%), 硫酸铁(Fe2(SO4)3, Fe含量为21%~23%), 1, 10-邻菲啰啉(C12H8N2·H2O, ≥99%)和盐酸羟胺(HONH3Cl, ≥98.5%)购买于天津市大茂试剂厂; 盐酸(HCl, 36%~38%)和氢氧化钾(KOH, ≥85%)购于广州化学试剂厂; 碳酸钠(Na2CO3, ≥99.8%)和碳酸氢钠(NaHCO3, ≥99.5%)购买于上海泰坦科技股份有限公司; 氢氧化钠(NaOH, >98%)购于福晨(天津)化学试剂有限公司.
2.2 含As(V)黄钾铁矾的合成含As(V)黄钾铁矾的合成主要参照Baron和Palmer(1996)的方法, 即将装有100 mL去离子水的烧杯置于油浴锅中加热, 直至水温为95 ℃, 加入14.1 g Fe2(SO4)3与5.6 g KOH, 再加入0.2 g Na2HAsO4·7H2O, 搅拌均匀溶解后, 在95 ℃油浴锅中持续搅拌加热4 h.悬浮液冷却静置后将沉淀物用去离子水充分清洗、离心、冷冻干燥, 经玛瑙研钵充分研磨后过200目筛, 存于干燥器中备用.
2.3 稳定性实验在稳定性实验中, 草酸盐储备液现配现用, 稀释至0、0.1、1和5 mmol·L-1, 用HCl或NaOH调节草酸盐溶液pH值至2.5和6.5.含As(V)黄钾铁矾在pH 2.5体系中, 随着反应的进行, 体系的pH值变化范围较小, 因此反应体系中未添加缓冲溶液; 在pH 6.5体系中, 加入0.05 mol·L-1 MES缓冲溶液.
本研究采用牺牲实验的方法.在一系列20 mL棕色色谱瓶中装入0.03 g含As(V)黄钾铁矾, 然后加入15 mL溶液(0、0.1、1和5 mmol·L-1的草酸盐溶液), 每个处理组设置3个平行样.将装有含As(V)黄钾铁矾悬浮液的棕色色谱瓶放入恒温摇床中, 在25 ℃下避光以150 r·min-1的速度振荡.在60 d的反应过程中, 在设定的时间间隔内定期取样, 取样后悬浮液离心进行固液分离, 离心机转速为8000 r·min-1, 时间为5 min.用注射器吸取上层清液经0.22 μm微孔滤膜过滤, 滤液置于4 ℃冰箱保存; 下层固体颗粒经冷冻干燥后进行固相分析.
2.4 分析方法与仪器用邻菲啰啉分光光度法测定溶液中的总Fe浓度, 波长为510 nm(UV-160A, Shimadzu, Japan). K+释放量采用火焰原子吸收光谱仪(Z-2700, Hitachi, Japan)测定; SO42-和草酸盐在溶液相中浓度采用离子色谱仪(Dionex ICS-1500, USA)测定; 总As含量采用原子荧光光谱仪(AFS-9130, Jitan)测定.
准确称取0.05 g初始合成的含As(V)黄钾铁矾溶解于HCl, 室温下静置消解24 h后, 取适量消解液稀释, 测定初始矿物中Fe、K、S和As的元素含量.
采用X射线衍射光谱(XRD, D8 ADVANCE, Bruker)确定矿物相组成和晶型结构; 采用傅里叶红外光谱(FTIR, VERTEX 33, Bruker)分析矿物表面官能团特征, 扫描范围为4000~400 cm-1; 利用X射线光电子能谱(XPS, Axis Ulra DLD, Krotos)对固相中Fe、As、O和C元素进行分析, 研究含As(V)黄钾铁矾反应前后各元素的成键状态和价态; 采用比表面分析仪(NOVA4200e, Netherlands)对黄钾铁矾颗粒进行比表面积分析测定.
3 结果与讨论(Results and discussion)3.1 含As(V)黄钾铁矾的合成和鉴定水热法合成含As(V)黄钾铁矾的XRD谱图如图 1所示, 经与标准谱图比对, 具有全部黄钾铁矾的特征峰, 并且没有出现多余的杂峰, 表明共沉淀的AsO43-没有改变黄钾铁矾的特征结构(Baron et al., 1996). Kendall等(2013)报道, 经XRD分析, 负载3.7%砷酸根的黄钾铁矾没有观察到矿物结构的改变.
图 1(Fig. 1)
 |
| 图 1 含As(V)黄钾铁矾在pH 2.5(a)和pH 6.5(b)时在不同初始草酸盐浓度下反应前后XRD谱图 Fig. 1XRD patterns of As(V)-bearing jarosite at pH 2.5 (a) and pH 6.5(b) before and after reaction in different initial oxalate concentrations |
含As(V)黄钾铁矾FTIR谱图如图 2所示.位于2900~3700 cm-1是O—H的伸缩振动特征峰, 1636 cm-1和1005 cm-1处的峰分别为HOH和O—H变形伸缩振动特征峰.矿物上SO42-分子伸缩振动特征峰主要包括两部分, 位于1187 cm-1和1087 cm-1是ν3(SO4)特征峰; 两个位于670 cm-1和630 cm-1的肩峰为ν4(SO4)特征峰. 575、513和477 cm-1特征峰对应的是FeO6八面体结构(Baron et al., 1996; Forray et al., 2014).从图 1和图 2可以看出, 本研究中合成含As(V)黄钾铁矾的XRD和FTIR特征峰与文献中报道的矿物特征峰一致, 表明此方法合成的含As(V)黄钾铁矾具有典型黄钾铁矾的结构特征.
图 2(Fig. 2)
 |
| 图 2 含As(V)黄钾铁矾在pH 2.5(a)和pH 6.5(b)时在不同初始草酸盐浓度下反应前后FTIR谱图 Fig. 2FTIR spectra of As(V)-bearing jarosite at pH 2.5 (a) and pH 6.5 (b)before and after reaction in different initial oxalate concentrations |
经过元素分析测定(表 1), K:Fe:S比例为0.95:2.56:2.02, 接近典型黄钾铁矾的1:3:2元素比例, 但K和Fe元素含量有少量缺失(周顺桂等, 2004).这是由于在矿物合成过程中, AsO43-取代矿物结构中的SO42-会引起电荷不平衡和结构扭曲, 从而导致K和Fe元素含量下降(Welch et al., 2007).
表 1(Table 1)
| 表 1 矿物组成及比表面积 Table 1 Components of synthetic As(V)-bearing jarosite and BET surface area | ||||||||||||||
表 1 矿物组成及比表面积 Table 1 Components of synthetic As(V)-bearing jarosite and BET surface area
| ||||||||||||||
3.2 草酸盐对含As(V)黄钾铁矾稳定性的影响3.2.1 草酸盐对总铁、钾和硫酸根离子释放的影响含As(V)黄钾铁矾与草酸盐混合后, 溶液相中总Fe、K+和SO42-的浓度迅速上升后趋于稳定, 这归因于含As(V)黄钾铁矾与草酸盐的快速反应(图 3).总Fe(图 3a)释放量随草酸盐浓度增大而增加, 在5 mmol·L-1草酸盐浓度条件下, 总Fe溶解量为反应体系中矿物总Fe含量的29.7%;空白对照处理中总Fe释放量仅为0.604 mmol·L-1. K+和SO42-(图 3b和3c)的溶解行为与总Fe相似, 在5 mmol·L-1草酸盐浓度体系中, 于第3 d达到最大值(分别为1.029 mmol·L-1和3.323 mmol·L-1), K+和SO42-在液相中的总含量分别为反应体系中对应其离子总含量的27.1%和13.7%;空白对照处理组中, K+和SO42-在反应过程中溶解量都分别低于0.4 mmol·L-1.这些结果都表明, 在pH 2.5条件下稳定存在的黄钾铁矾, 在草酸盐加入的情况下, 草酸盐能促进矿物中总Fe、K+和SO42-在溶液中的积累, 并且草酸盐浓度越高, 对含As(V)黄钾铁矾的溶解促进作用越强.
图 3(Fig. 3)
 |
| 图 3 含As(V)黄钾铁矾在pH 2.5时不同初始草酸盐浓度条件下, 溶液相中总Fe(a)、K+(b)、SO42-(c)和总As(d)浓度变化 Fig. 3Changes in aqueous-phase concentrations of total Fe(a), K+(b), SO42-(c) and total As(d) in different initial oxalate concentrations at pH 2.5 during reaction |
草酸为小分子二元羧酸, 易于与矿物表面的Fe(III)结合形成Fe(III)-草酸盐络合物, 从而吸附在黄钾铁矾表面, 与腐植酸等大分子相比, 草酸空间位阻较小, 形成的络合物更加稳定(Wang et al., 2017). Fe(III)-草酸盐络合物的形成是矿物溶解的第一步和关键(Yu et al., 2013). Paris和Desboeufs(2013)认为溶解速率取决于矿物表面形成的络合物数量.反应过程中溶液相草酸盐浓度如图 5a所示.在初始浓度为0.1 mmol·L-1草酸盐体系中, 在反应一开始草酸盐就完全吸附到矿物表面; 在初始浓度为1和5 mmol·L-1的草酸盐体系中, 在反应第一天内, 分别有29.4%和27.5%草酸盐吸附到矿物上, 这与总Fe、K+和SO42-在溶液相中浓度快速增加相对应(图 3).
图 5(Fig. 5)
 |
| 图 5 与含As(V)黄钾铁矾在pH 2.5(a)和pH 6.5(b)条件下反应过程中溶液相草酸盐浓度百分比 Fig. 5Percent of oxalate concentration in aqueous phase after reaction with As(V)-bearing jarosite at pH 2.5 (a) and pH 6.5(b) |
含As(V)黄钾铁矾在pH 6.5时的溶解过程与在pH 2.5条件下不相同. 图 4b和4c显示, 草酸盐的存在抑制含As(V)黄钾铁矾K+和SO42-的释放, 并且随着草酸盐浓度增加, 抑制作用逐渐增强. 图 5b表明, 溶液中的草酸盐在反应过程中逐渐吸附在矿物表面, 在5 mmol·L-1草酸盐体系中, 60 d后溶液相仅剩22.2%草酸盐, 而在0.1 mmol·L-1和1 mmol·L-1草酸盐体系中, 液相中的草酸盐全部吸附到矿物上.在反应过程中, 含As(V)黄钾铁矾的表面活性位点或孔隙对于草酸盐积累而言是饱和的, 造成一定数量的草酸盐不会直接与矿物表面位点或孔隙结合, 而是形成有机酸多分子层, 覆盖在矿物表面的溶解位点, 从而使K+和SO42-的释放受空间位阻限制(Chen et al., 2014).在5 mmol·L-1草酸盐体系中, 反应60 d后K+释放量为反应体系中矿物总钾含量的60.2%, SO42-释放量为反应体系中矿物总硫酸根含量的28.7%, 总Fe释放量为反应体系中矿物总铁含量的6.3%.反应后期, 溶液中总Fe浓度不断下降, 这是由于在高pH条件下溶液相的Fe(III)通过水解或沉淀作用重新吸附到矿物表面或形成二次矿物(Kendall et al., 2013), 并且新形成的矿物结构不含K+和SO42-结合的活性位点, 使黄钾铁矾中的K+和SO42-不断释放到溶液中并累积(Smith et al., 2006); 同时当pH升高时, 黄钾铁矾表面所带正电荷减少, 对矿物表面的SO42-吸附性能降低, SO42-与OH-的交换作用增强(李君菲等, 2018), 这些因素导致K+和SO42-浓度在pH 6.5条件下高于pH 2.5.
图 4(Fig. 4)
 |
| 图 4 含As(V)黄钾铁矾在pH 6.5时不同初始草酸盐浓度条件下, 溶液相中总Fe(a)、K+(b)、SO42-(c)和总As(d)浓度变化 Fig. 4Changes in aqueous-phase concentrations of total Fe(a), K+(b), SO42-(c) and total As(d) in different initial oxalate concentrations at pH 6.5 during reaction |
3.2.2 草酸盐对共沉淀As(V)迁移的影响在pH 2.5体系中, 溶液中总As含量(图 3d)在加入草酸盐后迅速增加, 并在12 h内达到最大值(4.341~40.829 μmol·L-1), 随后总As浓度随时间略有下降, 反应结束时, As释放量为0.480~31.726 μmol·L-1; 空白对照组中总As释放量在60 d的实验期间一直很少, 低于反应体系中矿物总砷含量的0.4%, 这是由于含As(V)黄钾铁矾表面结合态As的解吸(Gadol et al., 2017).前期总As溶解量增加是由于草酸盐促进含As(V)黄钾铁矾的溶解, 使矿物结构中的As被释放出来, 甚至草酸盐在一定程度上能取代结合的As(Mikutta et al., 2014); 随着溶解过程的进行, 含As(V)黄钾铁矾暴露出更多的活性位点, 有助于As的重新吸附(Yu et al., 2013), 但大部分砷仍以溶解态形式存在, 这是由于草酸盐抑制释放的砷酸根的再吸附, 从图 3和图 5可以看出, 溶液相草酸盐浓度明显高于释放的砷酸根浓度, 更加有利于草酸盐竞争含As(V)黄钾铁矾表面的活性位点.吸附竞争作用是由于草酸盐与砷酸根具有相同的吸附机理, 砷酸根通过形成双齿单核、双齿双核和单齿单核络合物吸附到铁矿物表面, 形成Fe—O—As配位键; 草酸盐也能与铁矿物表面的Fe结合, 形成Fe—O—C配位键, 所以草酸盐和砷酸根同时竞争含As(V)黄钾铁矾表面的Fe原子, 竞争吸附导致As重新吸附到矿物相上更加困难(Yu et al., 2013; 钟松雄等, 2017).
在pH 6.5条件下, 释放的As(图 4d)几乎全部重新吸附在固体相上.溶液中的总As浓度, 除5 mmol·L-1草酸盐体系, 都低于反应体系中矿物总As含量的0.1%;在5 mmol·L-1草酸盐溶液中, 总As溶解量在第6 d达到最大值(12.529 μmol·L-1), 随后减少到0.270 μmol·L-1. As释放量在pH 6.5体系中少于pH 2.5, 这是由于在高pH值下, Fe(III)水解导致次生矿物的生成, 有利于As重新吸附(Ren et al., 2018). As的吸附位点可能在次生矿物生长位置, 但高浓度草酸盐对As吸附有一定的抑制作用(Flynn et al., 2018).
3.2.3 含As(V)黄钾铁矾晶相转化产物表征含As(V)黄钾铁矾相转变产物的XRD谱图如图 1所示.鉴定结果表明, 共沉淀了AsO43-的黄钾铁矾在pH 2.5条件下, 经与草酸盐反应60 d后(图 1a), 矿物组成仍是黄钾铁矾, 只是矿物特征峰峰强变弱, 说明矿物发生一定程度的溶解.在pH 6.5条件下(图 1b), 产物XRD谱图上出现纤铁矿与针铁矿特征峰, 表明有纤铁矿与针铁矿的生成(Karimian et al., 2017); 并且在5 mmol·L-1草酸盐体系下, 产物仍存在少量黄钾铁矾特征峰, 表明高浓度草酸盐对矿物溶解有一定的抑制作用, 与图 4b和4c相对应.含As(V)黄钾铁矾在草酸盐体系中的溶解、重结晶过程在不同pH条件下呈现较大的差异(图 3和图 4), 并且随着pH上升, 黄钾铁矾会向更稳定的晶型转变(Karimian et al., 2017; Fan et al., 2019b).
含As(V)黄钾铁矾重结晶产物的FTIR谱图(图 2)显示, 在pH 2.5条件下, 经60 d反应后(图 2a), 草酸盐体系产物的FTIR谱图与反应前黄钾铁矾谱图相比无明显变化, 说明在此条件下, 矿物结构没有改变, 仅发生溶解反应.在pH 6.5反应体系中(图 2b), FTIR谱图显示有新特征峰生成以及一些原始矿物官能团特征峰减弱甚至消失, 表明矿物结构发生改变.
位于2900~3700 cm-1的O—H特征峰峰强明显减弱, 且向低波长段偏移; 位于1636 cm-1的HOH伸缩振动特征峰增强并向高波长段偏移; 产物仍保留黄钾铁矾原始的O—H变形伸缩振动特征峰, 但峰强减弱, 且在0.1 mmol·L-1草酸盐体系中, 产物特征峰迁移至1019 cm-1.这表明草酸盐与含As(V)黄钾铁矾表面的—OH络合(Fan et al., 2019b).当溶液草酸盐浓度为5 mmol·L-1时, 出现1415 cm-1新特征峰, 表明草酸盐的COO-官能团通过配体交换机制与矿物表面Fe(III)形成羧酸盐金属键(Chen et al., 2014). ν3(SO4)和ν4(SO4)特征峰依然存在, 但强度明显减弱, 表明在反应过程中黄钾铁矾结构中SO42-流失; 在1122 cm-1处出现新特征峰, 这是由于SO42-可能以外圈络合物的形式吸附在新形成的矿物相上, 从而导致与黄钾铁矾相似的FTIR谱带(Wan et al., 2018).在887 cm-1和796 cm-1处出现的肩峰为针铁矿结构中的Fe—O—H, 表明在反应过程中, 有针铁矿生成(Smith et al., 2006), 并且在1 mmol·L-1草酸盐体系中, 产物的Fe—O—H特征峰峰强最强, 与XRD结果(图 1b)一致. FeO6八面体结构对应的特征峰消失, 这表明含As(V)黄钾铁矾结构的坍塌.
3.2.4 含As(V)黄钾铁矾元素形态分析为进一步探讨含As(V)黄钾铁矾在转化过程中各元素的存在形式和As的价态变化, 采用光电子能谱对反应前后固体样品的Fe 2p、C 1s、O 1s和As 3d结合能进行测定, 采用XPS PEAK 41软件对XPS谱图进行分峰拟合(图 6). Fe 2p3/2和Fe 2p1/2的结合能分别为711.9 eV和725.8 eV, 结合能均高于710 eV, 表明在初始矿物中铁以Fe(III)形式存在.对Fe 2p3/2进行分峰分析(图 6a), 拟合谱图得Fe(III)、Fe(III)-SO4和Fe(OH)O特征峰, 分别对应的电子结合能为713.9、711.9和711.1 eV(Fan et al., 2019b); 同时, Fe 2p卫星峰的结合能为718.9 eV(Ji et al., 2019).反应60 d后在转化产物的Fe 2p3/2谱图中新增加了FeOOH和FeO特征峰(图 6b), FeOOH特征峰的出现表明固相转化产物含有针铁矿组分(Fan et al., 2019b).
图 6(Fig. 6)
 |
| 图 6 含As(V)黄钾铁矾在pH 6.5时5 mmol·L-1草酸体系下反应前后Fe 2p(a~b), O 1s(c~d), C 1s(e~f)和As 3d(g~h)的XPS谱图 Fig. 6XPS of Fe 2p(a~b), O 1s(c~d), C 1s(e~f), As 3d(g~h) for As(V)-bearing jarosite before and after reaction with 5 mmol·L-1 oxalate at pH 6.5 |
从反应后样品O 1s的XPS谱图中可以清晰观察到有C=O、Fe(OH)O和Fe2O3(图 6d)特征峰(Xie et al., 2017). C 1s分峰结果显示了四种碳键的存在(图 6f), 分别为C=O、C—O、C—OH和C—C(Mikutta et al., 2014), XPS的C 1s谱图显示矿物表面C(in atom%)的相对含量增加7.7%, 表明在反应过程中草酸盐吸附在铁矿物表面, 使铁矿物表面含碳量增加, 这与FTIR结果一致.
将反应前后As 3d的XPS谱图进行比较, 确定As(V)价态是否发生变化.反应前(图 6g), As 3d结合能为45.7 eV和44.4 eV; 反应后(图 6h), As 3d的电子结合能为45.8 eV和44.8 eV.这些特征峰均属于As(V)—O(Ji et al., 2019; Pozdnyakov et al., 2016), 表明在反应过程中矿物相的As(V)没有被还原, 未转变为剧毒的As(III)化合物.
3.3 机理推测本研究主要探讨了在草酸盐介导下含As(V)黄钾铁矾的溶解、重结晶和As的迁移行为.由FTIR和XPS分析可知, 草酸盐可与黄钾铁矾表面的Fe(III)络合, 从而加速含As(V)黄钾铁矾的溶解和转化.双齿配体草酸盐可与铁矿物表面的Fe(III)活性位点结合形成双齿单核、双齿双核、单齿单核络合物(Wang et al., 2017), 促使Fe原子与O原子之间的键极化, 破坏Fe—O键的稳定性并降低Fe溶出的活化能, 从而形成相对较强的可溶性[≡FeIII(C2O4)n](2n-3)-表面络合物(Ren et al., 2018), 一部分表面络合物脱离矿物固相进入溶液相, 同时一部分被草酸盐络合的Fe(III)释放到溶液中, 如式(1)~(3)所示, 而游离的草酸盐能与矿物表面的另一个Fe(III)原子再次结合形成络合物, 草酸盐从矿物表面上的一个位置通过溶液传质到另一个位置的过程, 能促进黄钾铁矾的溶解(Gadol et al., 2017).发生溶解的表面区域暴露了先前共沉淀在矿物结构中的As; 同时As能以Fe为桥接物与草酸盐结合, 形成As-Fe-草酸盐络合物, 从而促进As释放(鲁宗杰等, 2017).在pH 2.5条件下, 草酸盐与As(V)竞争矿物表面的活性位点, 导致As(V)难以重新吸附到矿物相上; 随着pH从2.5上升到6.5, Fe(III)通过水解和沉淀作用, 生成的次生矿物针铁矿与纤铁矿具有更大的比表面积和更加丰富的≡Fe—OH基团(Fan et al., 2019a), 能够有效固定游离的As(V), 二次矿物的形成能控制溶解态Fe和As的再分配.
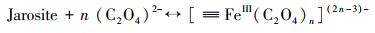 | (1) |
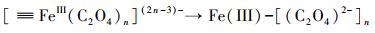 | (2) |
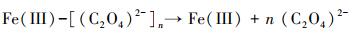 | (3) |
4 结论(Conclusions)本研究采用牺牲实验的方法探究草酸盐对含As(V)黄钾铁矾稳定性和共沉淀As迁移的影响, 结果表明初始草酸盐浓度与pH均对反应过程有作用.在pH 2.5条件下, 草酸盐通过与矿物表面的Fe(III)结合形成可溶性强络合物, 促进含As(V)黄钾铁矾的溶解, 并且草酸盐初始浓度越高, 溶解促进作用越明显, 并伴随着共沉淀的As(V)释放到溶液中.在pH 6.5体系中, 草酸盐促进含As(V)黄钾铁矾的相转变, 并且在草酸盐初始浓度为1 mmol·L-1时, 促进作用最强.经过XRD和XPS分析, 确定针铁矿与纤铁矿为主要产物并且能有效控制Fe和As的迁移; 同时在反应过程中As(V)并未转变为毒性更强的As(III).反应过程可概括为配体促进黄钾铁矾溶解、重结晶以及新生成的针铁矿与纤铁矿固定游离态的As.
因此在酸性矿山环境中, 如果想在低pH条件下控制砷污染, 应避免引入外源有机酸等, 保持系统的稳定性, 从而有利于在低pH环境下生成的铁矿物(如黄钾铁矾、施氏矿物等)吸附、共沉淀砷; 如果难以保持系统的稳定性, 则将pH调节至偏中性, 该条件有利于针铁矿、水铁矿、纤铁矿等生成, 能有效固定游离态砷.
参考文献
| Baron D, Palmer C D. 1996. Solubility of jarosite at 4~35℃[J]. Geochimica et Cosmochimica Acta, 60(2): 185-195. DOI:10.1016/0016-7037(95)00392-4 |
| Chen C, Dynes J J, Wang J, et al. 2014. Properties of Fe-organic matter associations via coprecipitation versus adsorption[J]. Environmental Science & Technology, 48(23): 13751-13759. |
| Eick M J, Peak J D, Brady W D. 1999. The effect of oxyanions on the oxalate-promoted dissolution of goethite[J]. Soil Science Society of America Journal, 63(5): 1133-1141. DOI:10.2136/sssaj1999.6351133x |
| Fan C, Guo C, Chen M, et al. 2019a. Transformation of cadmium-associated schwertmannite and subsequent element repartitioning behaviors[J]. Environmental Science and Pollution Research, 26(1): 617-627. DOI:10.1007/s11356-018-3441-9 |
| Fan C, Guo C, Zeng Y, et al. 2019b. The behavior of chromium and arsenic associated with redox transformation of schwertmannite in AMD environment[J]. Chemosphere, 222: 945-953. DOI:10.1016/j.chemosphere.2019.01.142 |
| Fang D, Wei S, Xu Y, et al. 2019. Impact of low-molecular weight organic acids on selenite immobilization by goethite:Understanding a competitive-synergistic coupling effect and speciation transformation[J]. Science of the Total Environment, 684: 694-704. DOI:10.1016/j.scitotenv.2019.05.294 |
| Flynn E D, Catalano J G. 2018. Influence of oxalate on Ni fate during Fe(II)-catalyzed recrystallization of hematite and goethite[J]. Environmental Science & Technology, 52(12): 6920-6927. |
| Forray F L, Smith A M L, Navrotsky A, et al. 2014. Synthesis, characterization and thermochemistry of synthetic Pb-As, Pb-Cu and Pb-Zn jarosites[J]. Geochimica et Cosmochimica Acta, 127: 107-119. DOI:10.1016/j.gca.2013.10.043 |
| Gadol H J, Flynn E D, Catalano J G. 2017. Oxalate-promoted trace metal release from crystalline iron oxides under aerobic conditions[J]. Environmental Science & Technology Letters, 4(7): 311-315. |
| Gao K, Jiang M, Guo C, et al. 2019. Reductive dissolution of jarosite by a sulfate reducing bacterial community:Secondary mineralization and microflora development[J]. Science of the Total Environment, 690: 1100-1109. DOI:10.1016/j.scitotenv.2019.06.483 |
| Ji Y, Luo W, Lu G, et al. 2019. Effect of phosphate on amorphous iron mineral generation and arsenic behavior in paddy soils[J]. Science of the Total Environment, 657: 644-656. DOI:10.1016/j.scitotenv.2018.12.063 |
| Karimian N, Johnston S G, Burton E D. 2017. Antimony and arsenic behavior during Fe(II)-induced transformation of jarosite[J]. Environmental Science & Technology, 51(8): 4259-4268. |
| Kendall M R, Madden A S, Elwood Madden M E, et al. 2013. Effects of arsenic incorporation on jarosite dissolution rates and reaction products[J]. Geochimica et Cosmochimica Acta, 112: 192-207. DOI:10.1016/j.gca.2013.02.019 |
| 刘奋武, 高诗颖, 王敏, 等. 2015. KOH对富铁富硫酸盐酸性环境中生物成因次生铁矿物合成的影响[J]. 环境科学学报, 35(2): 476-483. |
| 李君菲, 谢莹莹, 党志, 等. 2018. 酸性矿山废水中铜在含铬施氏矿物上的吸附及其对矿物溶解与相转变的影响[J]. 环境科学学报, 38(8): 3138-3145. |
| 鲁宗杰, 邓娅敏, 杜尧, 等. 2017. 江汉平原高砷地下水中DOM三维荧光特征及其指示意义[J]. 地球科学, 42(5): 771-782. |
| Mikutta R, Lorenz D, Guggenberger G, et al. 2014. Properties and reactivity of Fe-organic matter associations formed by coprecipitation versus adsorption:Clues from arsenate batch adsorption[J]. Geochimica et Cosmochimica Acta, 144: 258-276. DOI:10.1016/j.gca.2014.08.026 |
| Paris R, Desboeufs K V. 2013. Effect of atmospheric organic complexation on iron-bearing dust solubility[J]. Atmospheric Chemistry and Physics, 13(9): 4895-4905. DOI:10.5194/acp-13-4895-2013 |
| Pozdnyakov I P, Ding W, Xu J, et al. 2016. Photochemical transformation of an iron(III)-arsenite complex in acidic aqueous solution[J]. Photochemical & Photobiological Sciences, 15(3): 431-439. |
| Reichard P U, Kretzschmar R, Kraemer S M. 2007. Dissolution mechanisms of goethite in the presence of siderophores and organic acids[J]. Geochimica et Cosmochimica Acta, 71(23): 5635-5650. DOI:10.1016/j.gca.2006.12.022 |
| Ren H, Ji Z, Wu S, et al. 2018. Photoreductive dissolution of schwertmannite induced by oxalate and the mobilization of adsorbed As(V)[J]. Chemosphere, 208: 294-302. DOI:10.1016/j.chemosphere.2018.05.187 |
| Savage K S, Bird D K, Ashley R P. 2000. Legacy of the California Gold Rush:Environmental geochemistry of arsenic in the Southern Mother Lode Gold District[J]. International Geology Review, 42(5): 385-415. DOI:10.1080/00206810009465089 |
| Simanova A A, Loring J S, Persson P. 2011. Formation of ternary metal-oxalate surface complexes on α-FeOOH particles[J]. The Journal of Physical Chemistry C, 115(43): 21191-21198. DOI:10.1021/jp2058707 |
| Smith A M L, Hudson-Edwards K A, Dubbin W E, et al. 2006. Dissolution of jarosite[KFe3(SO4)2(OH)6] at pH 2 and 8:Insights from batch experiments and computational modelling[J]. Geochimica et Cosmochimica Acta, 70(3): 608-621. DOI:10.1016/j.gca.2005.09.024 |
| Strobel B W. 2001. Influence of vegetation on low-molecular-weight carboxylic acids in soil solution-a review[J]. Geoderma, 99(3): 169-198. |
| Wan J, Guo C, Tu Z, et al. 2018. Microbial reduction of Cr(VI)-loaded schwertmannite by Shewanella oneidensis MR-1[J]. Geomicrobiology Journal, 35(9): 727-734. DOI:10.1080/01490451.2018.1455767 |
| Wang Z, Fu H, Zhang L, et al. 2017. Ligand-promoted photoreductive dissolution of goethite by atmospheric low-molecular dicarboxylates[J]. The Journal of Physical Chemistry A, 121(8): 1647-1656. DOI:10.1021/acs.jpca.6b09160 |
| Welch S A, Christy A G, Kirste D, et al. 2007. Jarosite dissolution I-Trace cation flux in acid sulfate soils[J]. Chemical Geology, 245(3/4): 183-197. |
| Xie Y, Lu G, Yang C, et al. 2018. Mineralogical characteristics of sediments and heavy metal mobilization along a river watershed affected by acid mine drainage[J]. Plos One, 13(1): e190010. |
| Xie Y, Lu G, Ye H, et al. 2017. Fulvic acid induced the liberation of chromium from CrO42--substituted schwertmannite[J]. Chemical Geology, 475: 52-61. DOI:10.1016/j.chemgeo.2017.10.031 |
| Yang X, Zhu M, Kang F, et al. 2015. Formation mechanism of a series of trigonal antiprismatic jarosite-type compounds[J]. Journal of Crystal Growth, 429: 49-55. DOI:10.1016/j.jcrysgro.2015.07.037 |
| Yu B, Jia S, Liu Y, et al. 2013. Mobilization and re-adsorption of arsenate on ferrihydrite and hematite in the presence of oxalate[J]. Journal of Hazardous Materials, 262: 701-708. DOI:10.1016/j.jhazmat.2013.09.010 |
| 周顺桂, 周立祥, 黄焕忠. 2004. 黄钾铁矾的生物合成与鉴定[J]. 光谱学与光谱分析, 24(9): 1140-1143. DOI:10.3321/j.issn:1000-0593.2004.09.033 |
| 钟松雄, 尹光彩, 何宏飞, 等. 2017. 不同铁矿物对水稻土砷的稳定化效果及机制[J]. 环境科学学报, 37(5): 1931-1938. |
