 , 陈龙1, 黄韬博1, 陶溪1, 孙丰宾1, 刘文1,2,3, 齐娟娟1,2
, 陈龙1, 黄韬博1, 陶溪1, 孙丰宾1, 刘文1,2,3, 齐娟娟1,2
1. 北京大学环境科学与工程学院, 水沙科学教育部重点实验室, 北京 100871;
2. 北京大学工程科学与新兴技术高精尖创新中心, 北京 100871;
3. 北京大学环境工程系北京市新型污水深度处理工程技术研究中心, 北京 100871
收稿日期: 2019-12-09; 修回日期: 2020-01-06; 录用日期: 2020-01-06
基金项目: 中国博士后科学基金面上项目(No.2019M650007);国家自然科学基金项目(No.21906001)
作者简介: 潘柏岳(1993-), 男, E-mail:1701214517@pku.edu.cn
通讯作者(责任作者): 齐娟娟, E-mail:qijuanjuan@pku.edu.cn
摘要:制备了以KNbO3为载体材料的Co(OH)2复合材料并对其进行了详细的表征,分析了材料的组成成分、组成形态进而确定了其为核壳结构形貌的KNbO3@Co(OH)2.利用合成的样品作为催化剂活化过一硫酸盐(peroxymonosulfate,PMS)来降解帕珠沙星(pazufloxacin,PZF),结果表明制备的催化剂对PZF的去除效率显著增加.讨论了不同初始PMS剂量对降解效率的影响,发现随着PMS增加可活化生成更多的硫酸根自由基(sulfate radicals,
 )和羟基自由基(hydroxyl radicals,HO·)来降解PZF,但继续增大PMS用量降解效率未见明显提升.酸性和中性pH值条件下利于反应活化PMS降解PZF,而碱性体系减缓反应,甚至强碱体系更易形成Co(OH)2沉淀不利于反应体系中活性组分CoOH+的形成,大大抑制了催化性能.此外,在体系中加入淬灭剂叔丁醇(tert-Butanol,TBA)或者乙醇(ethanol,ETOH)进行自由基的淬灭实验,结果表明
)和羟基自由基(hydroxyl radicals,HO·)来降解PZF,但继续增大PMS用量降解效率未见明显提升.酸性和中性pH值条件下利于反应活化PMS降解PZF,而碱性体系减缓反应,甚至强碱体系更易形成Co(OH)2沉淀不利于反应体系中活性组分CoOH+的形成,大大抑制了催化性能.此外,在体系中加入淬灭剂叔丁醇(tert-Butanol,TBA)或者乙醇(ethanol,ETOH)进行自由基的淬灭实验,结果表明 自由基为体系降解PZF过程中主要贡献的自由基,而HO·自由基的贡献较少.催化剂具有较好的稳定性5次循环之后仍能在10 min之内完全去除PZF.本研究提出了新的思路为制备其他载体的Co(OH)2核壳结构提供参考依据,同时将该催化剂结合高级氧化技术应用到水体新兴有机污染物净化领域具有很好的应用前景.
自由基为体系降解PZF过程中主要贡献的自由基,而HO·自由基的贡献较少.催化剂具有较好的稳定性5次循环之后仍能在10 min之内完全去除PZF.本研究提出了新的思路为制备其他载体的Co(OH)2核壳结构提供参考依据,同时将该催化剂结合高级氧化技术应用到水体新兴有机污染物净化领域具有很好的应用前景.关键词:核壳结构KNbO3@Co(OH)2过一硫酸盐(PMS)硫酸根自由基帕珠沙星
Core-shell KNbO3@Co(OH)2 preparation and peroxymonosulfate activation for degradation of pazufloxacin
PAN Po-Yueh1
, CHEN Long1, HUANG Taobo1, TAO Xi1, SUN Fengbin1, LIU Wen1,2,3, QI Juanjuan1,21. The Key Laboratory of Water and Sediment Sciences, Ministry of Education, College of Environmental Sciences and Engineering, Peking University, Beijing 100871;
2. The Beijing Innovation Center for Engineering Science and Advanced Technology(BIC-ESAT), Peking University, Beijing 100871;
3. Beijing Engineering Research Center for Advanced Wastewater Treatment, Department of Environmental Engineering, Peking University, Beijing 100871
Received 9 December 2019; received in revised from 6 January 2020; accepted 6 January 2020
Abstract: In this study, KNbO3 was used to support Co(OH)2, and the composite material KNbO3@Co(OH)2 was fabricated into a core-shell structure based on the morphology and composition characterization. The synthesized material was applied as a heterogeneous catalyst to realize efficient activation of peroxymonosulfate (PMS) for pazufloxacin (PZF) removal. The effects of different initial PMS dosages on the degradation were investigated. It was found that with the increase of PMS, the generation of sulfate radicals (
 ) and hydroxyl radicals (HO·) were produced, leading to the increase of the degradation efficiency, but excessive PMS was not benefit to further PZF removal. The optimal pH for degradation were carried out under weak acidic or neutral pH conditions, while alkaline condition could slow down the reaction. Even higher alkaline were more likely to form Co(OH)2, which were adverse to the formation of the active component CoOH+, thus inhibiting reaction. Besides, quenching experiments by adding the tert-Butanol (TBA) or ethanol (ETOH), the results indicated that
) and hydroxyl radicals (HO·) were produced, leading to the increase of the degradation efficiency, but excessive PMS was not benefit to further PZF removal. The optimal pH for degradation were carried out under weak acidic or neutral pH conditions, while alkaline condition could slow down the reaction. Even higher alkaline were more likely to form Co(OH)2, which were adverse to the formation of the active component CoOH+, thus inhibiting reaction. Besides, quenching experiments by adding the tert-Butanol (TBA) or ethanol (ETOH), the results indicated that  radicals were predominant radicals. After five cycles, the catalyst was still good, as complete PZF removal was also achieved within 10 min. Finally, a new strategy for preparing the core-shell structure of Co(OH)2 by other carriers. The catalyst combined the advanced oxidation processes would have great application potential in the removal of emerging organic pollutants in water.
radicals were predominant radicals. After five cycles, the catalyst was still good, as complete PZF removal was also achieved within 10 min. Finally, a new strategy for preparing the core-shell structure of Co(OH)2 by other carriers. The catalyst combined the advanced oxidation processes would have great application potential in the removal of emerging organic pollutants in water.Keywords: core-shell structureKNbO3@Co(OH)2PMSsulfate radicals (
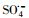 )pazufloxacin
)pazufloxacin1 引言(Introduction)帕珠沙星(pazufloxacin, PZF)为喹诺酮类抗生素之一, 具有抗菌活性强、抗菌谱广对革兰氏阳性菌和革兰氏阴性菌均有抗菌作用(Heeb et al., 2011), 与其他抗菌药物无交叉耐药, 副作用小等优点, 被广泛应用于临床医学、畜牧、水产养殖等领域的疾病防治(Ling et al., 2008; Li et al., 2018).但是随着帕珠沙星药物的广泛使用, 其在人体和动物体内吸收效率较低, 且在体内代谢较差, 半衰期较长, 最后通过代谢排泄到水体环境中.此外, 其他的生产途径如市政废水、养殖废水、医疗废水等排放的PZF逐渐成为不可忽视的环境问题.我国地表水、土壤、沉积物中均检测到ng·L-1~μg·L-1级别的抗生素, 这对长期暴露于低浓度的环境水生生物来说可能会发生抗性基因的变异(Cai et al., 2018), 而通过生物蓄积作用, 对生态系统和人体健康存在潜在风险, 亟须引起高度重视(Chen et al., 2019).目前来说传统的废水处理工艺并不能对其有效地去除.因此, 开发环境友好的帕珠沙星去除技术显得尤为重要.
基于硫酸根自由基(
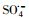

众所周知, 铌酸钾(KNbO3)材料(Grinberg et al., 2013; Grabowska, 2016)是一种钙钛矿类型的材料, 由于具有优越的物理化学性能、光催化活性、低毒性、良好的生物相容性、反应活性高、性能稳定等特点而受到研究者的广泛关注(Lan et al., 2011; Wang et al., 2013).基于KNbO3二阶非线性光学系数大、光照条件下性能稳定和丰富的晶相结构(导带具有空的d轨道)等特点, 使其在光催化去除水体污染物领域的研究被相继报道(Lan et al., 2011; Wang et al., 2013; Grabowska, 2016).如能结合KNbO3优良的光催化性能并协同钴系材料活化PMS体系, 将会实现快速去除水体污染物, 而本论文只考虑了单一影响因素即KNbO3和钴系材料复合后活化PMS去除水体污染物的研究, 后续会考虑光催化协同活化PMS去除水体污染物.因此, KNbO3可作为钴系的载体材料, 且由于表面富含大量的羟基基团, 使得钴离子与羟基形成羟基钴, 更利于实现KNbO3和钴系材料复合后活化PMS降解水体有机污染物.
基于此, 本研究制备了以KNbO3为载体材料的Co(OH)2复合材料并对其进行了详细的表征, 分析了材料的组成成分、组成形态确定了其为核壳结构形貌的KNbO3@Co(OH)2.利用制备的样品作为催化剂活化过一硫酸盐(PMS)来降解帕珠沙星(PZF), 探讨了不同初始PMS剂量、pH值对PZF降解效率的影响, 通过自由基淬灭实验了解体系主要贡献的自由基, 初步材料循环稳定性, 污染物矿化率及体系PMS的利用率等, 以期为后续其他钴系核壳材料的制备及水体新兴有机污染物净化领域提供理论参考依据.
2 材料与方法(Materials and methods)2.1 实验材料五氧化二铌购于国药集团化学试剂有限公司; PMS(peroxymonosulfate)购于Alfa Aesar公司; 氢氧化钠、氢氧化钾、碘化钾、高氯酸、六水合硝酸钴、乙醇(EtOH)、叔丁醇(TBA)均购于西陇科学股份有限公司; 五水硫代硫酸钠购于北京通广精细化工公司; 甲醇和乙腈(HPLC级)购于EMD Millipore公司; 甲酸购于上海阿拉丁生化科技股份有限公司; 实验所使用去离子水购于中镜科仪技术有限公司.
2.2 核壳结构KNbO3@Co(OH)2的制备① 首先制备立方体形貌的铌酸钾采用典型的水热法合成, 具体步骤如下:将86 g KOH溶于60 mL水中磁力搅拌, 此时体系放出大量的热待冷却至室温, 加入0.6 g五氧化二铌, 继续搅拌12 h, 转入带有不锈钢外层的聚四氟乙烯反应器中, 在150 ℃下保持72 h.反应结束自然冷却至室温后过滤洗涤沉淀至上清液pH接近中性, 过滤样品, 置于60 ℃烘箱中烘干得到样品A.
② 合成烷氧基钴(Zhao et al., 2014)作为Co(OH)2的前驱体, 将10 mmol·L-1 Co(NO3)2·6H2O溶解于60 mL异丙醇中, 然后添加16 mL丙三醇, 搅拌30 min至完全溶解, 转移到不锈钢外套的聚四氟乙烯反应器中, 并在180 ℃下加热6 h.加热结束自然冷却至室温后, 反应后沉淀物经乙醇多次洗涤, 过滤样品置于60 ℃烘箱烘干, 得到前驱体样品B.
③ 将0.1 g A和0.1 g B放入研钵一起研磨(加入烷氧基钴B号样品可避免体系中直接加入Co(NO3)2·6H2O会迅速形成沉淀, 不利于钴源均匀的生长在KNbO3A号样的表面)将混合后的粉体加入到聚四氟乙烯内衬中, 随后加入20 mL去离子水搅拌均匀, 将内衬转移到100 mL反应釜中进行第二次水热, 水热温度160 ℃保持3 h, 反应后样品烘干即为核壳结构KNbO3@Co(OH)2样品.
2.3 样品的表征样品通过环境扫描电镜(Environmental scanning electron microscopy, ESEM, FEI Quanta 250 FEG), 场发射扫描电镜(Field-emission SEM, 操作电压15 kV, Merlin Compact, ZEISS, 德国)及能谱分析EDS (Energy Dispersive Spectrometer)来进行样品微观形貌的观察和元素分析.透射电子显微镜(Transmission electron microscopy, TEM)和高分辨率透射电镜(High-resolution TEM, HRTEM)的型号为Tecnai 30 FEG透射电子显微镜(FEI, 美国, 操作电压300 kV)观察所制备样品的微观组成形态, 进而对样品晶体结构进行鉴定.X射线衍射(XRD)来进行表征, 仪器使用日本Rigaku公司的DMAX-2400, 在40 kV和100 mA条件下对材料扫描, 扫描衍射角度(2θ)为5°~70°, 扫描时长为20 min; 材料中的元素组成以及各元素的存在形态通过X射线光电子能谱(XPS)进行表征, 所使用的设备为Kratos Analytical Ltd生产的Axis Ultra多功能电子能谱仪, 结合能通过样品C1s峰位固定在284.8 eV来进行校准.钴离子的浓度测定采用电感耦合等离子体原子发射光谱(ICP-AES, Prodigy, Leeman, 美国)进行检测.
2.4 活化PMS降解PZF的实验活化PMS降解PZF的实验在200 mL烧杯中进行, 将1 mL浓度为1 mmol·L-1 PZF加入到100 mL水中搅拌, 随后加入100 μL浓度为100 mmol·L-1 PMS, 在磁力搅拌器上持续搅拌, 除了考察溶液初始pH值的影响时需使用0.1 mol·L-1 NaOH和HClO4将溶液调至特定pH值, 其他体系初始值pH值均调为7.加入10 mg催化剂, 开始反应并计时.在设定的时间点取样, 分别从反应装置中提取1 mL水样, 并立即通过0.22 mm有机尼龙滤膜过滤至样品瓶, 且样品瓶预先加入0.1 mL浓度为100 mmol·L-1 Na2SO3溶液, 以淬灭PMS产生的残余自由基.
淬灭剂实验:为了研究不同活性自由基对PZF降解的影响贡献, 在加入材料之前先提前分别加入50 mmol·L-1叔丁醇(tert-Butanol, TBA)和50 mmol·L-1乙醇(ethanol, ETOH), 以分别淬灭羟基自由基(HO·)和所有自由基(HO·和
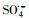
2.5 PZF测定方法及条件PZF浓度测定通过高效液相色谱(High Performance Liquid Chromatography, HPLC, Agilent 1260 Infinity II, USA)进行测定.进样量20 μL, 流速1 mL·min-1, 通过Agilent Zorbax SB-Aq(4.6 mm × 250 mm, 5 μm)液相色谱柱进行分离, 柱温箱温度控制在30 ℃, 流动相为70%甲醇和30% 0.1%甲酸水溶液, UV检测器波长为244 nm, 检测时间7 min.总有机碳TOC分析仪(Total Organic Carbon, 岛津, 日本)分析污染物的矿化效率.
2.6 反应动力学模型一般而言, 催化剂-PMS体系降解有机污染物其降解规律符合准一级反应动力学模型, 因此本研究利用核壳结构KNbO3@Co(OH)2材料活化PMS降解PZF也采用该模型来研究其降解规律, 模型如式(1)所示(Wang et al., 2017).
 | (1) |
2.7 体系中剩余PMS的测定分别反应时间点取5 mL样品加入0.5 g碘化钾, 充分摇晃后静置30 min.并经由紫外可见分光光度计(UV-1800, 岛津, 日本), 以空白参比在352 nm波长处测定吸光度值, 从标准曲线上查得试样中的PMS含量, 参考Lai课题组的工作(Zhang et al., 2019)来进行测定.
3 结果与讨论(Results and discussion)3.1 催化剂的表征3.1.1 ESEM和元素扫描分析利用环境扫描电镜(ESEM)对合成的样品进行表征.纯的KNbO3样品合成方法为制备的A号样品直接进行第③步的二次水热不加入钴源(即不加入B号样)所得样品, 从图 1a可以看出合成的KNbO3为小长方体形貌, 表面光滑, 分散性较好, 尺寸为400 ~ 500 nm左右.加入制备的烷氧基钴作为钴源, 以制备的长方体形貌的KNbO3作为载体材料, 发现制备的复合材料仍为长方体形貌, 但其表面不光滑变得粗糙, 粒径大小约为500 ~ 600 nm, 如图 1b~1c所示.进一步对其进行元素扫描分析, 如图 2a~2d所示, 从图中可看出Nb元素和O元素分布均匀含量较高, 呈现的形貌较为清晰, 而Co元素含量较少且主要附着在长方体的表面, 说明本研究合成了KNbO3和钴系的复合材料, 为了进一步证明附着在KNbO3表面钴的存在形式及壳层厚度需对样品结构进行详细的微观表征.
图 1(Fig. 1)
 |
| 图 1 KNbO3(a)、核壳结构KNbO3@Co(OH)2(b)及对应的单个核壳结构(c)的ESEM图片 Fig. 1ESEM images of KNbO3(a) KNbO3@Co(OH)2with core-shell structure (b) and a close-up of one typical single crystal (c) |
图 2(Fig. 2)
 |
| 图 2 元素扫描分析图(a.KNbO3@Co(OH)2的ESEM图片, b.Nb-La线系, c.O-Ka线系, d.Co-Ka线系) Fig. 2Element mapping analysis of (a.ESEM image of KNbO3@Co(OH)2, b.Nb-La, c.O-Ka, d.Co-Ka) |
3.1.2 TEM分析对核壳结构的样品进行透射电镜(TEM)分析如图 3a~3c所示.通过第二次水热的方法, 以烷氧基钴为钴源在第二次水热过程中缓慢水解重结晶, 使钴离子在KNbO3长方体周围生长, 进而包覆在其表面形成核壳结构的KNbO3@Co(OH)2形貌.图 3a给出了单个的KNbO3周围包覆Co(OH)2的图片可清晰分辨出其核壳结构, 核壳结构形貌的长方体尺寸约为500 nm, 与观察的ESEM结果图 1c相吻合.进一步对边界部分进行放大观察, 验证其生长的情况, 采用高倍HRTEM进行观察, 如图 3b~3c所示, 浅灰色虚线方框部分的放大图, 发现包覆在立方体周围的物质具有非常清晰的晶格衍射条纹说明在KNbO3边界处生长了结晶性比较好的Co(OH)2, 同时发现KNbO3尺寸仍为400~500 nm, 而其表层的Co(OH)2壳层厚度为几十到100 nm左右, 后续可通过调控钴源的添加量和反应时间等影响因素对其壳层厚度进行精准调控, 可深入研究不同壳层厚度的Co(OH)2对有机污染物降解性能的影响.
图 3(Fig. 3)
 |
| 图 3 单个核壳结构KNbO3@Co(OH)2的透射电镜(TEM)图片(a)及对应的单个核壳结构的局部放大TEM(b)和高倍透射电镜(HRTEM)图片(c) Fig. 3TEM image of KNbO3@Co(OH)2core-shell structure (a), local enlarged TEM image (b), and the corresponding high resolution TEM (HRTEM) (c) |
3.1.3 XRD衍射分析为了确定合成的3种材料的组成成分、晶体结构(晶型)、晶系等本研究对样品做了粉体X射线衍射分析, 图 4a给出了3种物质的XRD分析谱图.图 4a中纯的KNbO3, 合成方法为制备的A号样品直接进行第③步的二次水热不加入钴源(即不加入B号样)所得的样品, 从图中可以看出所制备的铌酸钾样品结晶性好, 具有非常尖锐的衍射峰, 这与斜方晶系KNbO3衍射峰相对应, 晶格参数分别为a=0.5695 nm, b=0.5721 nm, c=0.3973 nm, 空间点群对应Cm2m(38)(标准卡片:JCPDS No.32-0822)(Lan et al., 2011), 几个主要的特征衍射峰2θ=22.08°、31.58°和45.01°分别对应斜方晶系KNbO3的(110)、(111)和(220)晶面衍射峰, 该结果与之前的报道结果相吻合(Lan et al., 2011).图 4a中制备的核壳结构KNbO3@Co(OH)2的样品, 与KNbO3相比其主要衍射峰出峰位未发生明显改变, 说明仍为斜方晶系, 但并未出现明显的钴的峰, 可能由于铌酸钾作为载体是体系的主要成分, 其峰强度尖锐, 把钴相关的峰掩盖; 也有可能以烷氧基钴为钴源在KNbO3体系中铌酸钾表面有大量的羟基基团, 材料溶于水之后适宜的pH值易于烷氧基钴的水解, 进而经过水热和重结晶的过程在其表面形成钴系的材料, 剩余的钴源可能在洗样过程中流失, 为进一步确定钴的存在形态对材料又做了XPS分析.图 4b钴的混合物(mix-Co)即Co(CO3)0.5(OH)·0.11H2O(缩写为Co-C, 标准卡片:JCPDS No.48-0083)(Zhao et al., 2014)和Co(OH)2(标准卡片:JCPDS No.02-0925)的混合物, 合成方法为制备的B号样品直接进行第③步的二次水热不加入长方体形貌的KNbO3(即不加入A号样)所得的样品.因此, 实验可通过调控钴源添加量进而获得不同配比、不同组分和壳层厚度的复合材料.
图 4(Fig. 4)
 |
| 图 4 KNbO3、KNbO3@Co(OH)2和mix-Co的X射线衍射(XRD)分析谱图(a)及混合钴材料(mix-Co)放大的XRD分析谱图(b) Fig. 4XRD patterns of KNbO3, KNbO3@Co(OH)2, and mix-Co (a); and high resolution XRD of mix-Co (b) |
3.1.4 XPS分析表面组成和化学状态通过X射线光电子能谱(XPS)来进一步分析核壳结构的KNbO3@Co(OH)2, 图 5a为KNbO3@Co(OH)2的材料的XPS全谱图, 没有杂质峰出现(结合能通过样品C1s峰位固定在284.8 eV来进行校准), 可清晰看到所含有的各种元素验证材料为复合材料.在高分辨率的XPS光谱中分析讨论了Nb 3d、Co 2p和O 1s轨道的峰.图 5b中Nb 3d5/2和Nb 3d3/2结合能分别对应206.61 eV和209.31eV, 这与报道的KNbO3的结合能相似(Wang et al., 2013).通过图 5c中Co 2p的高分辨XPS分析进一步分析了复合材料中的钴的存在形态.高分辨的Co 2p光谱由于自旋-轨道劈裂成2p3/2和2p1/2两个部分, 分别观察到Co 2p3/2轨道峰对应的结合能781.01和Co 2p1/2轨道峰对应的结合能796.81 eV, 由于二价钴其价电子结构特点, 在2p主峰高结合能(约5~6.8 eV)处出现较强的震激(shake-up)伴峰(此为二价钴离子特有属性)分别对应Co的2p3/2和2p1/2轨道峰结合能为785.71 eV和802.41 eV, 说明合成的Co(OH)2中钴的存在价态为正二价(Yang et al., 2010), 与之前报道的Co 2p结合能Co2+一致.通过文献可知纯的KNbO3中在530 eV处O 1s的结合能对应于材料的晶格氧(Lan et al., 2011), 而对KNbO3@Co(OH)2的复合材料中O 1s轨道峰进行分峰拟合了解其存在形态和表面羟基基团的变化, 如图 5d所示, 在531.31 eV处的O 1s结合能对应吸附氧的存在形态和Co(OH)2样品中的羟基基团(Co-OH)中氧的结合能(金属氢氧化物中氧的结合能位于531~532eV, 与文献报道的Co(OH)2的O 1s出峰位置基本一致, 确定本研究合成的材料为Co(OH)2)(Yang et al., 2007; Yang et al., 2010; Wan et al., 2017), 对于结合能在529.50 eV处出现的肩峰可能归因于Co(OH)2中晶格氧(O2-)(Liang et al., 2018), KNbO3中存在的晶格氧(Lan et al., 2011), 或者CoOOH存在的O1s的氧物种(Liang et al., 2018).由于Yang等报道了Co/TiO2体系中利用Co2+与TiO2表面的羟基结合形成钴(Co-OH)实现高效去除污染物的相关工作(Yang et al., 2007), 后续实验预计缩小钴源的添加量, 找到二者最佳配比, 使得Co2+和KNbO3表面的羟基结合形成羟基钴(Co-OH), 尽量减少外层Co(OH)2的生成即实现壳层厚度可控.因此, 通过上述表征说明成功制备了核壳结构KNbO3@Co(OH)2的样品.
图 5(Fig. 5)
 |
| 图 5 KNbO3@Co(OH)2的XPS谱图(a.样品的全谱图, b.Nb 3d, c.Co 2p, d.O1s) Fig. 5XPS spectra of KNbO3@Co(OH)2 (a.survey spectrum, b.Nb 3d spectrum, c.Co 2p spectrum, d.O1s spectrum) |
3.2 催化剂活化PMS降解PZF性能研究3.2.1 不同材料降解PZF不同材料活化过一硫酸盐降解帕珠沙星的研究结果见图 6.本研究探讨了只有PMS和污染物PZF存在的情况, 发现PZF几乎没有降解, 说明单纯的PMS不能降解去除帕珠沙星.考虑纯的KNbO3单独活化PMS降解PZF, 发现对降解PZF的效率为3% ~ 4%可认为对反应体系没有太大影响.进而考察合成的KNbO3@Co(OH)2材料在没有PMS参加的情况下10 min以内对PZF吸附效果, 结果表明材料对于污染物的吸附效率在5%以内, 可忽略材料对污染物的吸附作用.由于钴系材料可以快速活化PMS降解有机污染物, 已经被很多课题组相继报道(Yang et al., 2009; Yuan et al., 2011), 因此本研究也考察了单独合成的混合钴系材料(mix-Co)对于PMS的催化效果, 从图中可以看出1 min之内降解达到约为73%, 延长时间到10 min, 最终检测的降解效率为75.47%, 后续几分钟并未有明显降解, 说明Co(OH)2中浸出的Co2+瞬间消耗完过一硫酸盐而不能形成持久释放去除有机污染物.因此, 加入制备的核壳结构的KNbO3@Co(OH)2的样品作为催化剂活化PMS降解PZF, 发现降解效果非常显著, 从图中可以看出在1 min之内PZF快速降解, 在1 min的时候, 降解效率为73.35%, 5 min之内降解去除率达到100%, 实现快速活化PMS降解去除PZF的目的.先前的研究表明, 羟基钴是整个系统的主要活性物质(Yang et al., 2007), 因此, KNbO3@Co(OH)2活化PMS的机理(Hu et al., 2016)可进行如下解释:
 | (2) |
 | (3) |
 | (4) |
 | (5) |
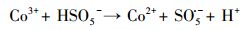 | (6) |
 | (7) |
 |
| 图 6 不同材料活化PMS去除PZF的影响(实验条件:[PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [催化剂]=0.1 g·L-1; pH=7.0 ± 0.2) Fig. 6Comparison of different catalyst-PMS systems on the removal of PZF(Experimental condition: [PZF]=10 μmol·L-1; [PMS]= 100 μmol·L-1; [catalyst]=0.1 g·L-1; pH=7.0±0.2) |
核壳结构的KNbO3@Co(OH)2中的Co(OH)2可产生有效活性物质CoOH+, 从而加快降解体系的反应速率.本研究以KNbO3作为载体材料, Co2+可与KNbO3上的羟基反应生成羟基钴的几率增加, 这从XPS中O 1s的分峰中发现有CoOOH和Co(OH)2结果相对应, 从而实现高效去除有机污染物.综上表明, 本研究制备的核壳结构的KNbO3@Co(OH)2的样品对于活化PMS降解PZF具有快速完全去除的特点.这或许因为催化剂KNbO3@Co(OH)2之间存在协同作用, 可在反应体系中高效活化PMS产生有效降解污染物的自由基实现对PZF完全降解.
3.2.2 PMS投加浓度的影响图 7给出了不同初始PMS添加量对降解PZF的影响, 图 7a给出了核壳结构的KNbO3@Co(OH)2的样品添加量为0.1 g·L-1时初始pH=7, 随着PMS的浓度从0到150 μmol·L-1, 1 min之内对应的降解效率分别为0、11.72%、44.26%、73.35%、71.21%;而10 min之内对于PZF的降解去除效率分别为5%、38.07%、75.29%、100%、100%.由于催化剂活化PMS体系降解有机污染物其降解规律一般符合准一级反应动力学模型, 因此本研究利用核壳结构KNbO3@Co(OH)2材料活化PMS降解PZF也采用该模型来研究其降解规律, 模型如式(1)所示, 如图 7b所示给出了5 min之内准一级反应动力学.不同初始浓度PMS添加量时, 一级反应速率从25 μmol·L-1的0.098 min-1增加到100 μmol·L-1的1.41 min-1, kobs增大了14.38倍.继续增加到150 μmol·L-1时, 反应速率kobs反而降低为1.20 min-1.对于整个反应体系的关键因素为羟基钴活化PMS产生硫酸根自由基, 随着PMS浓度的增加降解效率明显加快, 这是因为增加反应体系中PMS的浓度可导致更多的PMS被活化生成

图 7(Fig. 7)
 |
| 图 7 初始PMS添加量对降解PZF的影响(a.降解效率, b.准一级反应动力学模拟)(实验条件:[PZF]=10 μmol·L-1; [PMS]=0、25、50、100、150 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; pH=7.0 ± 0.2) Fig. 7Effect of initial PMS dosage on the removal of PZF (a.degradation efficiency, b.the corresponding pseudo-first-order kinetic model fitting) (Experimental condition: [PZF]=10 μmol·L-1; [PMS]= 0, 25, 50, 100, 150 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; pH=7.0 ±0.2) |
3.2.3 初始pH值的影响由于pH会影响有机物的存在形态和PMS主要自由基的类型, 因此对体系活化PMS降解去除PZF起着至关重要的作用.设计实验初始的pH值从3.00到11.00, 探索初始pH值对PZF降解的影响, 如图 8所示.在图 8a中, 随着pH从3.00增加到11.00, PZF的去除率先增加后减小, 5 min之内降解效率分别为59.79%、100%、100%、68.26%、2.89%.pH为5.00和7.00时, 10 min之内PZF的去除效率均为100%, pH值为9.00和11.00时, 去除效率显著下降.对其5 min之内反应动力学进行准一级动力学模拟, 如图 8b所示, 随着pH值从3.00增加到7.00, 反应速率kobs明显增加, 然后当pH从7.00进一步增加到11.00时kobs急剧减小, 这与以前的研究一致, Co2+引发的PMS的最佳pH区间在4.00和9.00之间(Chan et al., 2009).以上结果表明, 弱酸性和中性条件更有利于催化反应的进行.这主要归因于以下几点:①不同体系的pH值会影响PMS的组成形态, 由于过一硫酸(H2SO5)的pKa1 < 0, pKa2=9.4(Xu et al., 2019), 而PZF的pKa1=5.7和pKa2=8.6 (Sousa et al., 2013).因此, 当溶液处于酸性和中性条件下
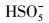
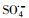
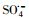


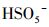



图 8(Fig. 8)
 |
| 图 8 初始pH值对PZF降解的影响(a.不同pH值对降解效率的影响, b.准一级反应速率, c.不同pH值下PZF的存在形态)(实验条件, [PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; pH=(3~11)± 0.2) Fig. 8Effect of initial pH on the degradation of PZF (a.degradation efficiency, b.the relationship between pH and the pseudo-first-order rate constant (kobs), c.existing form of PZF at different pH) (Experimental condition: [PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; pH=(3~11)± 0.2) |
3.2.4 自由基淬灭实验过渡金属离子活化过一硫酸盐(PMS)可能会产生HO·、






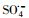

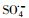
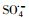

图 9(Fig. 9)
 |
| 图 9 自由基淬灭实验(a.添加不同淬灭剂对降解效率的影响, b.准一级反应动力学模拟)(实验条件:[PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; [TBA] = 50 mmol·L-1; [EtOH] = 50 mmol·L-1; pH=7.0 ± 0.2) Fig. 9Effect of different scavenger (a.degradation efficiency, b.the corresponding pseudo-first-order kinetic model fitting) (Experimental condition: [PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [KNbO3@Co(OH)2]=0.1 g·L-1; [TBA] = 50 mmol·L-1 or [EtOH] = 50 mmol·L-1; pH=7.0 ± 0.2) |
3.2.5 材料的回用实验、污染物矿化率及PMS的利用率对KNbO3@Co(OH)2催化剂的回用实验和稳定性进行了评价, 在最佳实验条件下做了5个循环的催化剂活化PMS去除PZF的重复实验.实验后的样品经过去离子水和乙醇多次洗涤, 然后放置于80 ℃烘箱中干燥1 h.做下一个循环时尽量保证添加的催化剂的量为0.1 g·L-1, 图 10a给出了5个循环材料的降解效率在3 min之内的降解效率从99%降至到第5次循环的78%, 虽然KNbO3@Co(OH)2催化剂的催化性能有所下降, 但10 min之内也能实现完全去除PZF.PZF的去除效率降低可能归因于以下两个方面:①由于样品为核壳机构的KNbO3@Co(OH)2, 外层的Co(OH)2可能在体系中部分被溶解或者脱落, 因为测试体系中Co2+的溶出率为7.8%.②去除PZF所产生的中间产物吸附在材料表面占据了催化剂反应的活性位点, 因此, 活化PMS产生的自由基减少, 导致降解效率有所下降(Zhang et al., 2019).此外, 为了了解KNbO3@Co(OH)2-PMS体系对PZF的降解的TOC去除率即污染物的矿化率, 对反应不同时间的溶液进行了TOC分析测试, 如图 10b所示.延长反应时间至60 min时, 测试TOC去除率为38%, 而当反应延长至2 h, TOC去除率约为45%, 矿化率并没有随着时间延长有显著的提升.原因可能是因为在反应之初PMS的浓度较高, 催化剂可活化PMS产生的自由基较多, 而随着反应的进行PMS被消耗浓度降低, 其对中间产物的矿化效率较低.为了进一步验证KNbO3@Co(OH)2催化剂的催化活性, 了解PMS在该体系中的利用率.通过不同反应时间点取样加入KI与体系中剩余的

图 10(Fig. 10)
 |
| 图 10 KNbO3@Co(OH)2活化PMS去除PZF的回用实验(a)、体系对PZF降解的TOC去除率(b)及反应体系中剩余的PMS(c)(实验条件:[PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [催化剂]=0.1 g·L-1; pH=7.0 ± 0.2) Fig. 10Recyclability of KNbO3@Co(OH)2 by PMS activation on the removal of PZF (a), the TOC removal efficiency of PZF (b) and residual PMS during the reaction system (c) (Experimental condition: [PZF]=10 μmol·L-1; [PMS]=100 μmol·L-1; [catalyst]=0.1 g·L-1; pH=7.0 ± 0.2) |
4 结论(Conclusions)1) 本研究制备了以KNbO3为载体材料的Co(OH)2复合材料并对其进行了ESEM, TEM, XRD和XPS等表征, 分析了材料的组成成分、组成形态进而确定了其为核壳结构形貌的KNbO3@Co(OH)2.
2) 利用制备的核壳结构KNbO3@Co(OH)2样品作为催化剂活化过一硫酸盐来降解帕珠沙星, 结果表明制备的催化剂对PZF的降解效率显著增加.探讨不同初始PMS剂量对降解效率的影响, 发现随着PMS增加可活化生成更多的

3) 酸性和中性pH值条件下利于反应活化PMS降解PZF, 而碱性体系减缓反应, 甚至强碱体系更易形成Co(OH)2沉淀不利于反应体系中活性组分CoOH+的形成, 大大抑制了催化性能.此外, 在体系中加入淬灭剂TBA和EtOH进行自由基的淬灭实验, 结果表明

4) 本研究提出了新的思路制备钴的核壳结构形貌为制备其他载体的钴系核壳材料提供参考依据, 同时将该催化剂结合高级氧化技术应用到水体新兴有机污染物净化领域具有很好的应用前景.
参考文献
| Cai C, Zhang H, Zhong X, et al. 2015. Ultrasound enhanced heterogeneous activation of peroxymonosulfate by a bimetallic Fe-Co/SBA-15 catalyst for the degradation of Orange II in water[J]. Journal of Hazardous Materials, 283: 70-79. DOI:10.1016/j.jhazmat.2014.08.053 |
| Cai Z, Dwivedi A D, Lee W N, et al. 2018. Application of nanotechnologies for removing pharmaceutically active compounds from water:development and future trends[J]. Environmental Science-Nano, 5(1): 27-47. DOI:10.1039/C7EN00644F |
| Chan K H, Chu W. 2009. Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate:Different cobalt counteranions in homogenous process and cobalt oxide catalysts in photolytic heterogeneous process[J]. Water Research, 43(9): 2513-2521. DOI:10.1016/j.watres.2009.02.029 |
| Chen D, Ma X, Zhou J, et al. 2014. Sulfate radical-induced degradation of Acid Orange 7 by a new magnetic composite catalyzed peroxymonosulfate oxidation process[J]. Journal of Hazardous Materials, 279: 476-484. DOI:10.1016/j.jhazmat.2014.06.004 |
| Chen Q, Chen L, Qi J J, et al. 2019. Photocatalytic degradation of amoxicillin by carbon quantum dots modified K2Ti6O13 nanotubes:Effect of light wavelength[J]. Chinese Chemical Letters, 30(6): 1214-1218. DOI:10.1016/j.cclet.2019.03.002 |
| Du Y, Ma W, Liu P, et al. 2016. Magnetic CoFe2O4 nanoparticles supported on titanate nanotubes (CoFe2O4/TNTs) as a novel heterogeneous catalyst for peroxymonosulfate activation and degradation of organic pollutants[J]. Journal of Hazardous Materials, 308: 58-66. DOI:10.1016/j.jhazmat.2016.01.035 |
| Grabowska E. 2016. Selected perovskite oxides:Characterization, preparation and photocatalytic properties-A review[J]. Applied Catalysis B-Environmental, 186: 97-126. DOI:10.1016/j.apcatb.2015.12.035 |
| Grinberg I, West D V, Torres M, et al. 2013. Perovskite oxides for visible-light-absorbing ferroelectric and photovoltaic materials[J]. Nature, 503(7477): 509-512. DOI:10.1038/nature12622 |
| Guan Y H, Ma J, Ren Y M, et al. 2013. Efficient degradation of atrazine by magnetic porous copper ferrite catalyzed peroxymonosulfate oxidation via the formation of hydroxyl and sulfate radicals[J]. Water Research, 47(14): 5431-5438. DOI:10.1016/j.watres.2013.06.023 |
| Heeb S, Fletcher M P, Chhabra S R, et al. 2011. Quinolones:from antibiotics to autoinducers[J]. Fems Microbiology Reviews, 35(2): 247-274. DOI:10.1111/j.1574-6976.2010.00247.x |
| Hu P, Long M. 2016. Cobalt-catalyzed sulfate radical-based advanced oxidation:A review on heterogeneous catalysts and applications[J]. Applied Catalysis B-Environmental, 181: 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| Lan J, Zhou X, Liu G, et al. 2011. Enhancing photocatalytic activity of one-dimensional KNbO3 nanowires by Au nanoparticles under ultraviolet and visible-light[J]. Nanoscale, 3(12): 5161-5167. DOI:10.1039/c1nr10953g |
| Li H, Zhang P, Liu Y, et al. 2012. Photophysical properties of gatifloxacin in aqueous solution by laser flash photolysis and pulse radiolysis[J]. Radiation Physics and Chemistry, 81(1): 40-45. |
| Li S, Shi W, Liu W, et al. 2018. A duodecennial national synthesis of antibiotics in China's major rivers and seas (2005-2016)[J]. Science of the Total Environment, 615: 906-917. DOI:10.1016/j.scitotenv.2017.09.328 |
| Liang Z, Yang Z, Huang Z, et al. 2018. Novel insight into the epitaxial growth mechanism of six-fold symmetrical beta-Co(OH)2/Co(OH)F hierarchical hexagrams and their water oxidation activity[J]. Electrochimica Acta, 271: 526-536. DOI:10.1016/j.electacta.2018.03.186 |
| Ling S K, Wang S, Peng Y. 2010. Oxidative degradation of dyes in water using Co2+/H2O2 and Co2+/peroxymonosulfate[J]. Journal of Hazardous Materials, 178(1/3): 385-389. |
| Ling X, Zhong W, Huang Q, et al. 2008. Spectroscopic studies on the interaction of pazufloxacin with calf thymus DNA[J]. Journal of Photochemistry and Photobiology B-Biology, 93(3): 172-176. DOI:10.1016/j.jphotobiol.2008.07.008 |
| Nie M, Yang Y, Zhang Z, et al. 2014. Degradation of chloramphenicol by thermally activated persulfate in aqueous solution[J]. Chemical Engineering Journal, 246: 373-382. DOI:10.1016/j.cej.2014.02.047 |
| 聂明华, 徐文丽, 张文静, 等. 2018. Fe2+和零价铁活化过一硫酸盐降解酸性橙7[J]. 环境科学学报, 38(10): 3997-4005. |
| 聂明华, 吴乐良, 余林兰, 等. 2018. 零价铁-过硫酸盐体系同时去除水中酸性橙7和磷[J]. 环境科学学报, 38(11): 4321-4332. |
| Pagano M, Volpe A, Mascolo G, et al. 2012. Peroxymonosulfate-Co(II) oxidation system for the removal of the non-ionic surfactant Brij 35 from aqueous solution[J]. Chemosphere, 86(4): 329-334. DOI:10.1016/j.chemosphere.2011.09.010 |
| Shi P, Su R, Zhu S, et al. 2012. Supported cobalt oxide on graphene oxide:highly efficient catalysts for the removal of orange II from water[J]. Journal of Hazardous Materials, 229: 331-339. |
| Sousa J, Alves G, Campos G, et al. 2013. First liquid chromatography method for the simultaneous determination of levofloxacin, pazufloxacin, gatifloxacin, moxifloxacin and trovafloxacin in human plasma[J]. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 930: 104-111. DOI:10.1016/j.jchromb.2013.04.036 |
| Tan C, Fu D, Gao N, et al. 2017. Kinetic degradation of chloramphenicol in water by UV/persulfate system[J]. Journal of Photochemistry and Photobiology a-Chemistry, 332: 406-412. DOI:10.1016/j.jphotochem.2016.09.021 |
| Tsitonaki A, Petri B, Crimi M, et al. 2010. In situ chemical oxidation of contaminated soil and groundwater using persulfate:A Review[J]. Critical Reviews in Environmental Science and Technology, 40(1): 55-91. DOI:10.1080/10643380802039303 |
| Wan S, Qi J, Zhang W, et al. 2017. Hierarchical Co(OH)F superstructure built by low-dimensional substructures for electrocatalytic water oxidation[J]. Advanced Materials, 29(28): 1700286. DOI:10.1002/adma.201700286 |
| Wang J, Wang S. 2018. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 334: 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
| Wang Q, Shao Y, Gao N, et al. 2017. Activation of peroxymonosulfate by Al2O3-based CoFe2O4 for the degradation of sulfachloropyridazine sodium:Kinetics and mechanism[J]. Separation and Purification Technology, 189: 176-185. DOI:10.1016/j.seppur.2017.07.046 |
| Wang R, Zhu Y, Qiu Y, et al. 2013. Synthesis of nitrogen-doped KNbO3 nanocubes with high photocatalytic activity for water splitting and degradation of organic pollutants under visible light[J]. Chemical Engineering Journal, 226: 123-130. DOI:10.1016/j.cej.2013.04.049 |
| 王莹, 魏成耀, 黄天寅, 等. 2017. 氮掺杂碳纳米管活化过一硫酸盐降解酸性橙AO7[J]. 中国环境科学, 37(7): 2583-2590. DOI:10.3969/j.issn.1000-6923.2017.07.021 |
| Xie P, Ma J, Liu W, et al. 2015. Removal of 2-MIB and geosmin using UV/persulfate:contributions of hydroxyl and sulfate radicals[J]. Water Research, 69: 223-233. DOI:10.1016/j.watres.2014.11.029 |
| Xu M, Li J, Yan Y, et al. 2019. Catalytic degradation of sulfamethoxazole through peroxymonosulfate activated with expanded graphite loaded CoFe2O4 particles[J]. Chemical Engineering Journal, 369: 403-413. DOI:10.1016/j.cej.2019.03.075 |
| Xu Y, Ai J, Zhang H. 2016. The mechanism of degradation of bisphenol A using the magnetically separable CuFe2O4/peroxymonosulfate heterogeneous oxidation process[J]. Journal of Hazardous Materials, 309: 87-96. DOI:10.1016/j.jhazmat.2016.01.023 |
| 徐闻欣, 吴俊峰, 邢俊明, 等. 2019. 羟胺强化亚铁活化过硫酸盐降解对乙酰氨基酚的研究[J]. 环境科学学报, 39(10): 3410-3417. |
| Yang J, Liu H, Martens W N, et al. 2010. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs[J]. Journal of Physical Chemistry C, 114(1): 111-119. DOI:10.1021/jp908548f |
| Yang Q J, Choi H, Al-Abed S R, et al. 2009. Iron-cobalt mixed oxide nanocatalysts:Heterogeneous peroxymonosulfate activation, cobalt leaching, and ferromagnetic properties for environmental applications[J]. Applied Catalysis B-Environmental, 88(3/4): 462-469. DOI:10.1016/j.apcatb.2008.10.013 |
| Yang Q, Choi H, Chen Y, et al. 2008. Heterogeneous activation of peroxymonosulfate by supported cobalt catalysts for the degradation of 2, 4-dichlorophenol in water:The effect of support, cobalt precursor, and UV radiation[J]. Applied Catalysis B-Environmental, 77(3/4): 300-307. DOI:10.1016/j.apcatb.2007.07.020 |
| Yang Q, Choi H, Dionysiou D D. 2007. Nanocrystalline cobalt oxide immobilized on titanium dioxide nanoparticles for the heterogeneous activation of peroxymonosulfate[J]. Applied Catalysis B-Environmental, 74(1/2): 170-178. |
| Yuan R, Ramjaun S N, Wang Z, et al. 2011. Effects of chloride ion on degradation of Acid Orange 7 by sulfate radical-based advanced oxidation process:Implications for formation of chlorinated aromatic compounds[J]. Journal of Hazardous Materials, 196: 173-179. DOI:10.1016/j.jhazmat.2011.09.007 |
| Zhang R, Wan Y, Peng J, et al. 2019. Efficient degradation of atrazine by LaCoO3/Al2O3 catalyzed peroxymonosulfate:Performance, degradation intermediates and mechanism[J]. Chemical Engineering Journal, 372: 796-808. DOI:10.1016/j.cej.2019.04.188 |
| 张晓峰, 林亲铁, 罗昊昱, 等. 2019. 海胆状钴铜双金属活化过硫酸钠降解土壤中六氯联苯[J]. 环境科学学报, 39(5): 1633-1638. |
| Zhao J, Zou Y C, Zou X X, et al. 2014. Self-template construction of hollow Co3O4 microspheres from porous ultrathin nanosheets and efficient noble metal-free water oxidation catalysts[J]. Nanoscale, 6(13): 7255-7262. DOI:10.1039/c4nr00002a |
