 , 刘海龙1
, 刘海龙1 , 吉力1, 李焕峰2
, 吉力1, 李焕峰21. 山西大学环境与资源学院, 太原 030006;
2. 山西省环境监测中心站, 太原 030002
收稿日期: 2018-05-07; 修回日期: 2018-09-07; 录用日期: 2018-09-07
基金项目: 国家自然科学基金(No.51179099,E090301);山西省重点研发计划项目(No.201603D321007);山西省科技攻关项目(No.20140313003-3);山西省留学回国人员科技活动择优资助项目(2014);山西省回国留学人员科研资助项目(No.2015-004)
作者简介: 陈璐(1996-), 女, E-mail:15735171925@163.com
通讯作者(责任作者): 刘海龙(1971—), 男, 博士, 教授, 主要研究方向为水处理机理及应用技术.E-mail: hlliu827@aliyun.com
摘要: 研究了低温条件下单独臭氧及MgO催化臭氧化降解水中氨氮的效率和特征,并对其反应机制分别进行了探讨.结果表明,pH是影响臭氧和催化臭氧化除氨的重要因素,不仅影响溶液中NH3与NH4+的比例和臭氧氧化氨氮的速率,还影响氧化产物种类,从而影响脱氮效果.10℃时,单独臭氧对水中氨氮的氧化降解效率随pH的升高而增大,pH≤9时整体降解效率不高,pH=9时仅为16.39%,而pH=10时达到41.77%.臭氧和·OH共同参与降解氨氮的过程.单独臭氧氧化氨氮生成氮气的选择性具有pH依赖性,并与Cl-密切相关.pH低(≤9)时,氨氮多以NH4+形态存在,O3与Cl-反应生成ClOx-(x=1、3),再氧化NH4+,从而生成气态产物N2或N2O.MgO在低温条件下具有很强的催化臭氧化降解氨氮的能力且温度升高有利于反应的进行,0、10、20℃时,MgO催化臭氧化氨氮的效率分别为77.53%、80.17%、91.26%.此过程中,·OH参与反应的程度低,一部分氨氮降解依靠ClOx-氧化NH4+,而氨氮降解的主要途径为O3对NH3的直接氧化.
关键词:氨氮臭氧MgO催化臭氧化机制
Mechanism of catalytic ozonation for degradation of ammonia nitrogen over MgO at low temperature
CHEN Lu1
, LIU Hailong1 , JI Li1, LI Huanfeng2 1. School of Environmental Sciences and Resources, Shanxi University, Taiyuan 030006;
2. Shanxi Environment Detection Central Station, Taiyuan 030002
Received 7 May 2018; received in revised from 7 September 2018; accepted 7 September 2018
Supported by the National Natural Science Foundation of China(No.51179099, E090301), the Key Research and Development Project of Shanxi Province(No.201603D321007), the Science and Technology Project of Shanxi Province(No.20140313003-3), the Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province(2014) and the Research Project Supported by Shanxi Scholarship Council of China(No.2015-004)
Biography: CHEN Lu(1996—), female, E-mail:15735171925@163.com
*Corresponding author: LIU Hailong, E-mail:hlliu827@aliyun.com
Abstract: The degradation efficiency and characteristics of ammonia in water by ozonation alone and ozonation catalyzed with MgO were studied under low temperature conditions. The reaction mechanisms were discussed, respectively. Results show that pH significantly influences the removal of ammonia by ozonation and catalytic ozonation, affecting not only the NH3 over NH4+ ratio in the solution but also the ozonation rate of ammonia as well as the types of oxidation products. At 10℃, the degradation efficiency of ammonia by ozonation alone increases with the increase of pH, which is relatively low at pH ≤ 9, only 16.39% at pH=9 while 41.77% at pH=10. Ammonia degradation is the result of direct molecular O3 and ·OH attack. And the selectivity to gaseous products is pH dependent in the presence of Cl-. At low pH(≤ 9), ammonia mainly exists in the form of NH4+. ClOx- (x=1, 3) generated by the reaction of O3 with Cl- reacts with NH4+ to produce gaseous products including N2 or N2O. MgO has a strong catalytic ability for ozonation to degrade ammonia at a low temperature, and the temperature increase helps to promote the degradation. 77.53%, 80.17% and 91.26% ammonia removal is gained at 0, 10 and 20℃, respectively. In the process, the contribution of ·OH to the ammonia removal is insignificant. The main degradation pathway for ammonia is direct ozone oxidation with the form of NH3 and a part of ammonia is degraded by the reaction of ClOx- with NH4+.
Keywords: ammoniaozoneMgOcatalytic ozonationmechanism
1 引言(Introduction)氨氮污染是水处理中普遍存在的问题.自然水体中的氨氮主要来源于石油化工、有色金属化学冶金、化肥、味精、肉类加工和养殖等工农业部门排放的废水(何岩等, 2008), 以及农业灌溉、动物排泄物和垃圾渗滤液等.水中的氨氮以铵根(NH4+)和游离氨(NH3)两种形式存在(Cheng et al., 2015), 其比例随水温和pH值的变化而变化.氨氮易被微生物氧化成硝酸盐氮和亚硝酸盐氮, 不仅会消耗水中的溶解氧, 且对水生生物和人体也有毒害作用(Tang et al., 2011);另外, 氨氮还是导致水体富营养化的因素之一.因此, 需严格控制水中氨氮含量.
目前, 处理水中氨氮的方法主要有离子交换法(Tang et al., 2011)、生物处理法和化学氧化法等.其中, 生物脱氮技术是去除原水中氨氮最有效、最经济的方法, 已被广泛应用, 但其对温度、pH、营养物质等条件敏感, 尤其在冬季低温条件下, 生物作用受到严重抑制(Khuntia et al., 2013), 从而导致生物脱氮效率较低, 因此, 低温控制氨氮的研究受到人们的关注.
化学氧化法是一种重要的低温除氨方法, 臭氧氧化法是其中之一.臭氧作为一种相对安全的消毒剂和氧化剂已被广泛用于水处理中, 其对氨氮有一定的氧化能力(Singer et al., 1975), 且低温时臭氧的溶解性和有效利用率较高, 因此, 臭氧氧化除氨有可能成为低温化学除氨的一种有效途径.O3及其产生的·OH与氨氮的反应方程式分别如式(1)~(2)(Singer et al., 1975)和式(3)~(6)(Chen et al., 2018)所示.
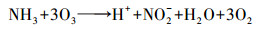 | (1) |
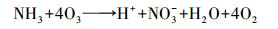 | (2) |
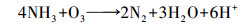 | (3) |
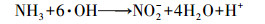 | (4) |
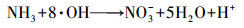 | (5) |
 | (6) |
2 材料与方法(Materials and methods)2.1 试剂和仪器所用试剂NH4Cl、NH4(SO4)2、MgCl2·6H2O、浓氨水、KI、叔丁醇等均为分析纯;所用水为实验室制超纯水;臭氧采用纯氧气源, 用自制双通道臭氧机制备.
2.2 催化剂(MgO)制备采用沉淀法制备MgO, 具体操作为:向1 mol·L-1 MgCl2·6H2O溶液中边搅拌边加入1 mol·L-1氨水至pH=10, 继续搅拌一段时间后静置20 min, 倒去上清液, 将沉淀过滤, 固体用纯水清洗3遍, 100 ℃烘干至恒重, 磨碎成粉末, 置于马弗炉中500 ℃焙烧4 h.
2.3 材料表征采用X射线粉末衍射仪(X′Pert PRO MPD)对MgO粉末进行结晶度和晶粒大小的分析, 具体参数为:Cu Kα, 波长1.540 nm, 管电压40 kV, 扫描角度5°~90°;采用X射线光电子能谱仪(ESCALAB250Xi)对MgO粉末进行表面元素组成分析, 具体参数为:Al Kα激发源, 发射功率250 W, 通过能量90 eV和35.75 eV分别用于宽程扫描和窄扫描, 能量分辨率0.8 eV, 灵敏度80 kCPS, 角分辨45°, 分析室真空度2.9×10-7 Pa, 实验得到的元素电子结合能以C 1s(285.0 eV)进行校正.
2.4 臭氧氧化实验臭氧发生器出口连接氨氮氧化反应瓶(250 mL, 含50 mg·L-1NH4Cl溶液), 开启磁力搅拌器使反应过程中臭氧与氨氮废水充分接触, 分别于0、15、30和60 min时取样测定氨氮、硝氮等含量;并连接KI吸收瓶处理尾气中的臭氧.
2.5 分析方法臭氧浓度测定采用靛蓝法(蒋丽春等, 2011), 氨氮含量测定采用纳氏试剂分光光度法(2010)、离子色谱法(Michalski et al., 2006), NO3--N含量测定采用紫外分光光度法(2007)、离子色谱法(Michalski et al., 2006).
3 结果与讨论(Results and discussion)3.1 pH对O3氧化氨氮的影响NH4Cl在水中的解离导致了NH4+和NH3之间的平衡, 具体方程可以表示为:
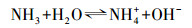 | (7) |
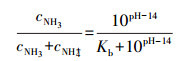 | (8) |
O3氧化氨氮的情况如图 1所示, 其中, 反应条件为:NH4Cl浓度50 mg·L-1, 溶液体积250 mL, T=10 ℃, 臭氧浓度4.56 mg·L-1, pH=5、6、7、8、9、10, 接触时间60 min.由图可知, 氨氮去除率随溶液初始pH的增大而增大, pH < 9时整体去除率较低, 在初始pH=5和6时, 去除率极低(< 3%), pH=9时去除率仅为16.39%, 而pH=10时去除率达到41.77%.O3和·OH与氨氮的反应(式(1)~(6))导致H+的生成, 因此, 初始pH=10、9、8、7的NH4Cl溶液在反应过程中pH都逐渐下降, 且因为反应程度不同而使pH降幅依次减小, 最终降至7.64、7.38、7.15、6.63, 反应速率也随之下降.因此可以认为, 尽管初始pH值不同, 当反应体系pH < 6时, O3不再继续氧化氨氮.
图 1(Fig. 1)
 |
| 图 1 pH对O3氧化氨氮的影响 Fig. 1Effect of pH on the ozonation of ammonia nitrogen |
氨氮在水中的氧化包括O3与氨氮的直接反应, 以及O3分解形成的·OH(其氧化电位为2.8 V, 高于O3氧化电位2.07 V(Enjarlis, 2014))与其进行的一系列链反应.Hoigné和Bader(1978)认为臭氧不能氧化NH4+, 只能氧化游离NH3.10 ℃时, pH=5、6、7、8、9、10的NH4Cl溶液中NH3浓度分别为0.0032、0.032、0.32、2.99、19.45、43.22 mg·L-1.随着pH的升高, NH3浓度增大, 提高了O3对其的氧化去除率.O3氧化NH3的速率见表 1.由表可知, 溶液初始pH越高, O3氧化NH3的速率越大.而在每个特定pH下, O3氧化NH3的速率随反应的进行均逐渐下降, 这与Singer和Zilli(1975)的研究结果吻合.pH越高, NH3浓度增大, 臭氧对其氧化速率越大.在每一特定pH下, 随反应进行, 溶液pH的逐渐下降导致游离NH3浓度进一步减小, 反应速率随之进一步下降.
表 1(Table 1)
| 表 1 不同初始pH下O3氧化NH3速率 Table 1 The degradation rate of NH3 by O3 oxidation at different initial pH | |||||||||||||||||||||||
表 1 不同初始pH下O3氧化NH3速率 Table 1 The degradation rate of NH3 by O3 oxidation at different initial pH
| |||||||||||||||||||||||
用叔丁醇作为·OH抑制剂探究是否有·OH参与反应, 结果见图 2, 其中, 反应条件为:NH4Cl浓度50 mg·L-1, 溶液体积250 mL, T=10 ℃, 臭氧浓度4.56 mg·L-1, pH=7、8、9、10, 接触时间60 min, 叔丁醇浓度20 mg·L-1.叔丁醇与·OH的反应速率常数为5×108 L·mol-1·s-1(Ma et al., 2000), 远远大于其与臭氧的反应速率常数k=0.03 L·mol-1·s-1(Hoigné et al., 1983), 从而作为自由基反应干扰物, 阻碍·OH与氨氮反应.由图 2可知, 加入抑制剂后, 不同pH下氨氮去除率均有明显下降, 且随pH升高, 去除率下降程度增大, 说明·OH参与了降解氨氮的反应, 而且pH越高, ·OH的参与程度越高.这是因为O3分解为·OH的速率与溶液pH有关(Elovitz et al., 2000), 较低pH下, ·OH生成量低;而在碱性条件下, OH-促进O3分解为活性·OH(钟理, 2000), ·OH量增大, 从而增大了降解氨氮的机会.
图 2(Fig. 2)
 |
| 图 2 不同初始pH下叔丁醇抑制臭氧氧化氨氮情况 Fig. 2Inhibitory effect of tert-butyl alcohol on ozonation of ammonia nitrogen at different initial pH |
3.2 O3氧化氨氮生成气态产物选择性分析对O3氧化氨氮进行产物分析(图 3)发现, 初始pH越低, 生成产物中硝氮的比例越低;在各个pH的反应体系中, 均未检测到NO2-的存在.Rosenthalh和Kruner(1985)认为, 在O3氧化体系中, 亚硝酸盐的存在会阻碍O3对氨氮的氧化.当体系中存在亚硝酸盐时, O3会优先氧化亚硝酸盐.O3对亚硝氮的氧化速率非常快, 在反应前几分钟就几乎被彻底氧化, 随着时间延长, 亚硝氮浓度并无增加.
图 3(Fig. 3)
 |
| 图 3 臭氧氧化NH4Cl和(NH4)2SO4生成硝氮的对比 Fig. 3Comparison of nitrate produced by ozonation of NH4Cl and (NH4)2SO4 |
将原液从NH4Cl换成(NH4)2SO4, 其余实验条件不变, 以探讨Cl-的作用, 对比结果见图 3.各个初始pH下, (NH4)2SO4溶液与O3反应生成的硝氮比例均比在相同条件下反应的NH4Cl溶液高, 生成产物几乎全部为硝氮, 由此说明Cl-对臭氧氧化氨氮反应的选择性起着至关重要的作用.以下对其作用机制进行了探讨.
Von Gunten(2003)研究认为, Cl-不能直接被O3氧化, 而是先由·OH将其氧化成Cl(+Ⅰ), 反应方程式为:
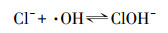 | (9) |
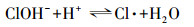 | (10) |
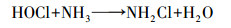 | (11) |
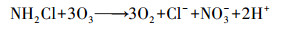 | (12) |
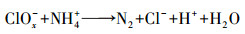 | (13) |
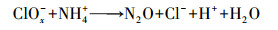 | (14) |
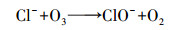 | (15) |
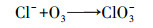 | (16) |
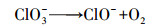 | (17) |
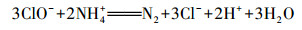 | (18) |
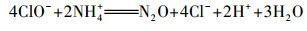 | (19) |
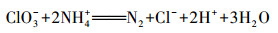 | (20) |
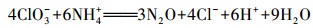 | (21) |
图 4(Fig. 4)
 |
| 图 4 臭氧氧化氨氮反应机制 Fig. 4Reaction mechanism of ammonia nitrogen oxidation by O3 |
3.3 催化臭氧化降解氨氮MgO催化臭氧化降解氨氮的效果如图 5所示, 其中, 反应条件为:NH4Cl浓度50 mg·L-1, 溶液体积250 mL, T=10 ℃, 臭氧浓度4.56 mg·L-1, pH=9, 催化剂投量1 g·L-1, 接触时间60 min.由图可知, MgO催化臭氧化氨氮的去除率随着反应的进行逐渐增大, 60 min时达到77.53%.反应速率在前30 min基本保持稳定;30~60 min之间速率略有下降, 应该是氨氮浓度因氧化去除而降低所致.催化反应的一个突出特征是反应结束时溶液pH升至10.32, 与单独O3氧化氨氮的情况(溶液pH随着反应进行而逐渐降低)明显不同.催化反应中MgO使溶液pH升高, 维持在10以上, 一方面增大了NH3的比例, 促进了O3对其直接氧化;另一方面促进了自由基的生成(钟理, 2000)及其对氨氮的降解, 从而提高了催化臭氧化去除氨氮的效率.
图 5(Fig. 5)
 |
| 图 5 MgO催化臭氧化降解氨氮效果 Fig. 5Degradation of ammonia nitrogen by catalytic ozonation over MgO |
3.3.1 氨氮降解机制氨氮被催化降解的途径可能有两种:一是直接催化氧化, 即催化O3直接氧化溶液中的NH3;二是间接催化氧化, 催化O3分解产生·OH与溶液中的氨氮发生反应(郭琳等, 2017).在低pH下, 以直接臭氧氧化为主, 而间接途径在碱性条件下由于OH-催化O3分解产生了大量的·OH而占优势(Agustina et al., 2005;Staehelin et al., 1985).对于不同的反应体系, 反应机制可能存在差异, 本研究针对MgO催化臭氧化降解氨氮的机制进行了探讨.
用叔丁醇作为·OH抑制剂探究是否有·OH参与反应, 结果见图 6, 其中, 反应条件为:NH4Cl/(NH4)2SO4浓度50 mg·L-1, 溶液体积250 mL, T=10 ℃, 臭氧浓度4.56 mg·L-1, pH=9, 催化剂投量1 g·L-1, 接触时间60 min, 叔丁醇浓度20 mg·L-1.加入抑制剂后, 氨氮去除率仅比对照组降低3.26%, 生成硝氮占降解氨氮的比例上升了4.52%, 说明·OH对催化臭氧化反应影响不大.这可能是由于催化剂表面经常起到自由基清除剂的作用(Lina, 2016), 所以由MgO存在时, ·OH浓度低于相同条件下催化剂不存在的情况, 使其不能发挥氧化氨氮的主要作用.
图 6(Fig. 6)
 |
| 图 6 叔丁醇对催化臭氧化NH4Cl与(NH4)2SO4的影响 Fig. 6Effects of tert-butyl alcohol on catalytic ozonation of NH4Cl and (NH4)2SO4 |
相同条件下, 用(NH4)2SO4溶液代替NH4Cl溶液验证Cl-是否参与催化臭氧化反应及其对产物选择性的影响(图 6).由图可知, (NH4)2SO4的氨氮去除率较NH4Cl仅降低了3.53%, 生成的硝酸盐氮比例却提高了19.65%, 达到96.95%, 即产物几乎全部为硝酸盐氮.Cl-显然对催化臭氧化除氨反应途径产生了重要的影响.在前述·OH对催化反应影响不大的基础上, 可以认为:使用NH4Cl反应时, 一部分氨氮通过O3与Cl-反应, 生成的ClOx-再氧化NH4+而降解, 从而生成的产物部分以N2或N2O形式存在, 降低了部分总氮.(NH4)2SO4溶液加入叔丁醇后催化臭氧化降解氨氮的效果几乎不受影响, 再次验证了催化反应中来自·OH的影响很小, 这是催化臭氧化反应与单独臭氧氧化反应(·OH发挥一定的降解氨氮的作用)的一大不同之处.
可见催化臭氧化除氨反应中, 氨氮降解的主要途径是O3直接氧化NH3, 仅有一部分氨氮是在其以NH4+的形态存在时被ClOx-氧化而降解.
3.3.2 MgO催化机制为了探究MgO的催化机制, 对其催化臭氧化反应前后分别进行了XRD(图 7)和XPS(图 8)分析.如图 7所示, 在2θ=42.9°和62.3°附近出现细长的尖峰, 表明实验中所制得的MgO结晶度较高, 而在其反应后, 结晶度受到一定程度影响.反应前后峰的位置没有明显变化, 说明反应过程中MgO晶格尺寸基本未改变.图 8a中XPS图谱主要显示Mg和O两种元素的峰及微弱的C、N杂峰, 表明MgO表面主要含有Mg和O两种元素, 而弱的C、N杂峰主要是MgO焙烧或者催化臭氧化反应过程中受到一定程度的污染所致.
图 7(Fig. 7)
 |
| 图 7 MgO催化臭氧化反应前后XRD分析图谱对比 Fig. 7XRD patterns of MgO before and after catalytic ozonation |
图 8(Fig. 8)
 |
| 图 8 MgO催化臭氧化反应前后XPS全谱分析对比(a)、Mg 1s(b)和Mg 2p(c)的XPS分析图谱对比 Fig. 8XPS patterns of MgO(a), Mg 1s(b), Mg 2p(c) before and after catalytic ozonation |
结合上述XRD和XPS图提出以下两种催化机制:①MgO使溶液pH升高, 催化O3直接氧化NH3的反应.从图 8b、8c可以看出, MgO在催化臭氧化反应之后, Mg 1s和2p电层的峰均显现一定程度的红移, 且峰面积和峰高发生改变, 表示在反应过程中与Mg2+结合的元素种类和数量发生了变化.因此, 关于MgO与pH的关系, 是由于MgO表面羟基化, 即晶格氧与水反应生成了OH-.MgO作为一种金属氧化物催化剂, 具有在水中表面羟基化的重要特性(徐贞贞, 2009), 且与水接触时其表面OH-均匀分布(印红玲等, 2003), 从而形成强碱性位.Tamura等(1999)用格氏试剂测得不同金属氧化物单位面积上的羟基物质的量基本相同, 据此认为金属氧化物表面的OH-形成不是因为其表面化学吸附水分子在金属离子(Lewis酸)上的离解(因为由此形成的OH-数目受金属价态的影响), 而是晶格氧(强碱)与水发生反应:
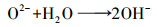 | (22) |
图 9(Fig. 9)
 |
| 图 9 氨氮在MgO表面的降解机制 Fig. 9Degradation mechanism of ammonia nitrogen on MgO surface |
图 10(Fig. 10)
 |
| 图 10 MgO吸附氨氮曲线 Fig. 10Adsorption of ammonia nitrogen by MgO |
3.4 温度对催化臭氧化氨氮的影响在整体低温条件下(0~20 ℃)探究了温度对催化臭氧化降解氨氮的影响, 结果见图 11, 其中, 反应条件为:NH4Cl浓度50 mg·L-1, 溶液体积250 mL, T=0、10、20 ℃, 臭氧浓度4.56 mg·L-1, pH=9, 催化剂投量1 g·L-1, 接触时间60 min.由图可知, 在0~20 ℃之间, 温度越高, MgO催化臭氧化降解氨氮的效率越高, 0、10、20 ℃时氨氮的去除率分别为77.53%、80.17%、91.26%.结果表明, 在0~20℃之间, 温度对氨氮的催化臭氧化作用有明显影响, 即使在低温(0~10℃)下, MgO催化臭氧化除氨仍保持了较高的去除率.同时, 温度越低臭氧在水中的溶解度越高, 有效利用率随之升高, 在一定程度上低温反而成为缩减臭氧氧化除氨成本的重要因素.可见MgO催化臭氧化可能成为冬季低温去除氨氮的备选方法之一.如何设计催化剂及提升将氨氮转化为氮气的比例等问题需进一步研究.
图 11(Fig. 11)
 |
| 图 11 温度对MgO催化臭氧化降解氨氮效果的影响 Fig. 11Effect of temperature on the degradation of ammonia nitrogen by catalytic ozonation over MgO |
4 结论(Conclusions)1) pH是影响臭氧除氨和催化臭氧化除氨的重要因素, 不仅会影响溶液中NH3与NH4+的比例和O3氧化氨氮的速率, 还会影响氧化产物的种类, 从而影响脱氮效果.
2) MgO具备很强的催化臭氧化降解氨氮的能力.氨氮被催化降解的主要途径为O3直接氧化NH3, 仅一部分通过O3氧化Cl-而生成的ClOx-(x=1、3)氧化NH4+, 进而生成气态产物N2和N2O实现.
3) MgO催化臭氧化的机制主要包括:①MgO部分溶解生成Mg(OH)2及表面强碱性位发生如O2-+H2O→2OH-的反应使溶液pH升高, 催化O3直接氧化NH3的反应;②MgO表面同时吸附NH3和O3在其表面发生反应, 反应完成后不断释放活性位点再进行下一次吸附降解.
4) 在0~20 ℃的温度区间, 温度越高, 氨氮的催化臭氧化去除率越高;即使在低温(0~10 ℃)下, MgO催化臭氧化除氨仍保持了较高的效率.
参考文献
| Agustina T E, Ang H M, Vareek V K. 2005. A review of synergistic effect of photocatalysis and ozonation on wastewater treatment[J]. Journal of Photochemistry and Photobiology C:Photochemistry Reviews, 6(4): 264–273.DOI:10.1016/j.jphotochemrev.2005.12.003 |
| Cheng C H, Yang F F, Ling R Z, et al. 2015. Effects of ammonia exposure on apoptosis, oxidative stress and immune response in pufferfish (Takifugu obscurus)[J]. Aquatic Toxicology, 164: 61–71.DOI:10.1016/j.aquatox.2015.04.004 |
| Chen Y N, Wu Y, Liu C, et al. 2018. Low-temperature conversion of ammonia to nitrogen in water with ozone over composite metal oxide catalyst[J]. Journal of Environmental Sciences, 66: 265–273.DOI:10.1016/j.jes.2017.04.032 |
| Enjarlis. 2014. Application of coagulation-advanced oxidation process by O3/GAC in the fan belt wastewater treatment[J]. Apcbee Procedia, 9: 145–150.DOI:10.1016/j.apcbee.2014.01.026 |
| Elovitz M S, Gunten U V, Kaiser H P. 2000. Hydroxyl radical/ozone ratios during ozonation processes.Ⅱ.The effect of temperature, pH, alkalinity, and DOM properties[J]. Ozone Science & Engineering, 22(2): 123–150. |
| 郭琳, 刘晨, 吴叶, 等. 2017. MgO催化臭氧氧化水中氨氮的研究[J]. 工业水处理, 2017, 37(6): 48–51. |
| Gunten U V. 2003. Ozonation of drinking water:Part Ⅱ.Disinfection and by-product formation in presence of bromide, iodide or chlorine[J]. Water Research, 37(7): 1469–1487.DOI:10.1016/S0043-1354(02)00458-X |
| 何岩, 赵由才, 周恭明. 2008. 高浓度氨氮废水脱氮技术研究进展[J]. 工业水处理, 2008, 28(1): 1–4. |
| Hoigné J, Bader H. 1978. Ozonation of water:kinetics of oxidation of ammonia by ozone and hydroxyl radicals[J]. Environmental Science & Technology, 12(1): 79–84. |
| Hoigné J, Bader H. 1983. Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅱ:dissociating organic compounds[J]. Water Research, 17(2): 185–194.DOI:10.1016/0043-1354(83)90099-4 |
| Ichikawa S I, Mahardiani L, Kamiya Y. 2014. Catalytic oxidation of ammonium ion in water with ozone over metal oxide catalysts[J]. Catalysis Today, 232: 192–197.DOI:10.1016/j.cattod.2013.09.039 |
| 蒋丽春, 唐绍明, 游青, 等. 2011. 靛蓝二磺酸钠褪色分光光度法测定水中臭氧[J]. 理化检验(化学分册), 2011, 47(2): 180–182. |
| Khuntia S, Majumder S K, Ghosh P. 2013. Removal of ammonia from water by ozone microbubbles[J]. Industrial & Engineering Chemistry Research, 52(1): 318–326. |
| Kuo C H, Yuan F, Hill D O. 1997. Kinetics of oxidation of ammonia in solutions containing ozone with or without hydrogen peroxide[J]. Industrial & Engineering Chemistry Research, 36(10): 4108–4113. |
| 刘永, 曹广斌, 蒋树义, 等. 2005. 冷水性鱼类工厂化养殖中臭氧催化氧化降解氨氮[J]. 中国水产科学, 2005, 12(6): 790–795. |
| Lin S H, Wu C L. 1996. Removal of nitrogenous compounds from aqueous solution by ozonation and ion exchange[J]. Water Research, 30(8): 1851–1857.DOI:10.1016/0043-1354(95)00329-0 |
| Lina M.2016.Cobalt oxide (Co3O4) as an active and selective catalyst for catalytic ozonation of ammonia nitrogen in water[D].Japan: Hokkaido University |
| Michalski R, Kurzyca I. 2006. Determination of nitrogen species (nitrate, nitrite and ammonia ions) in environmental samples by ion chromatography[J]. Polish Journal of Environmental Studies, 15(1): 5–18. |
| Ma J, Graham N J D. 2000. Degradation of atrazine by manganese-catalysed ozonation-influence of radical scavengers[J]. Water Research, 34(15): 3822–3828.DOI:10.1016/S0043-1354(00)00130-5 |
| Nawrocki J. 2013. Catalytic ozonation in water:Controversies and questions.Discussion paper[J]. Applied Catalysis B:Environmental, 142: 465–471. |
| Rosenthal H, Kruner G. 1985. Treatment efficiency of an improved ozonation unit applied to fish culture situations[J]. Ozone Science & Engineering, 7: 179–190. |
| Singer P C, Zilli W B. 1975. Ozonation of ammonia in wastewater[J]. Water Research, 9(2): 127–134.DOI:10.1016/0043-1354(75)90001-9 |
| 沈阳市环境监测中心站.2010.HJ 535-2009.水质氨氮的测定纳氏试剂分光光度法[S].北京: 中国环境科学出版社 |
| Schroeder J P, Croot P L, Von D B, et al. 2011. Potential and limitations of ozone for the removal of ammonia, nitrite, and yellow substances in marine recirculating aquaculture systems[J]. Aquacultural Engineering, 45(1): 35–41.DOI:10.1016/j.aquaeng.2011.06.001 |
| Staehelin J, Hoigne J. 1985. Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions[J]. Environmental Science & Technology, 19(12): 1206–1213. |
| Tang D Y, Zheng Z. 2011. Study on ammonia nitrogen adsorption from low concentration wastewater by modified zeolite and its desorption[J]. Chinese Journal of Environmental Engineering, 5(2): 293–296. |
| Tamura H, Tanaka A, Mita K Y, et al. 1999. Surface hydroxyl site densities on metal oxides as a measure for the ion-exchange capacity[J]. Journal of Colloid and Interface Science, 209(1): 225–231.DOI:10.1006/jcis.1998.5877 |
| 徐贞贞.2009.过渡金属羟基氧化物催化臭氧氧化水中痕量PCNB的研究[D].哈尔滨: 哈尔滨工业大学https://max.book118.com/html/2016/0316/37800392.shtm |
| 印红玲, 谢家理, 杨庆良, 等. 2003. 臭氧在金属氧化物上的分解机理[J]. 化学研究与应用, 2003, 15(1): 1–5. |
| 中华人民共和国国家环境保护总局.2007.HJ/T 346-2007.水质硝酸盐氮的测定紫外分光光度法(试行)[S].北京: 中国环境科学出版社 |
| 钟理. 2000. 臭氧湿式氧化氨氮的降解过程研究[J]. 中国给水排水, 2000, 16(1): 14–17. |
