
论文页面
高血压以体循环动脉血压增高、血管张力持续增加为主要特征,是导致心脑血管疾病和慢性肾功能衰竭等疾病的主要危险因素。尽管对高血压的发病机制进行了大量的实验研究,但其确切发病机制仍未十分清楚。因此,探索高血压发病机制是当今生命科学及医学领域的重要科学问题。
NO是人体内一个发现的重要气体信号分子,内皮源性NO是一种强效的血管扩张剂,对机体心血管系统稳态的维持起着重要作用。当NO的生物合成量减少或者被灭活时,可致血管壁的舒张功能的减低,是高血压发病的重要因素。内源性SO2以L-半胱氨酸为底物,由AAT催化生成。本课题组首次提出了内源性SO2作为心血管调节的第四种气体信号分子,在调节心血管功能与结构方面具有重要意义。
机体是一个复杂的体系,不同生物活性物质家系成员之间如肽类活性物质、氨基酸、气体信号分子之间的相互作用在血管调节中发挥重要的调节作用。然而,在高血压形成中气体信号分子的整合调控作用及其机制还不清楚。
为了探索高血压形成中气体信号分子SO2与NO在VEC与VSMC间通讯模式,阐明内源性SO2在内源性NO缺乏所致高血压发生中的补偿作用及意义,该课题组研究人员发现L-NAME处理的小鼠血浆NO含量显著降低,SO2含量显著升高,血压明显升高,血管重构加重。在此基础上再应用AAT抑制剂HDX抑制SO2生成,进一步加重了血管重构和血压升高。另外VECs和VSMCs共培养结果显示,与scramble组相比,在与eNOS敲低的VECs共培养的VSMCs中胶原蛋白表达明显增加;在此基础上,对VSMCs给予HDX干预后VSMCs中胶原蛋白表达进一步增加,说明内源性SO2体系作为内源性NO体系损伤的代偿性防御系统拮抗血管重构,拮抗高血压。
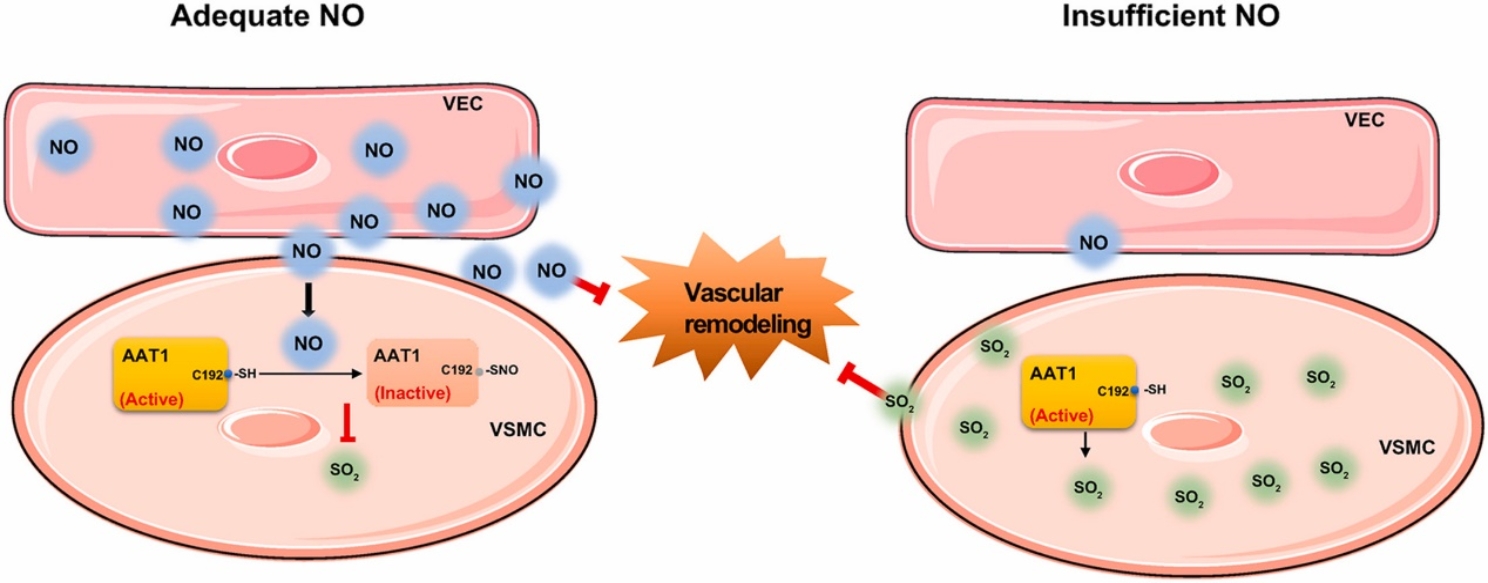
内源性SO2体系作为NO体系损伤的内源性防御系统,抑制血管重构
为了研究VEC来源的NO对VSMC来源SO2体系的作用机制,金红芳教授带领研究人员采用VECs与VSMCs共培养细胞模型,发现VECs来源的NO不影响VSMCs中AAT1蛋白的表达,但NO降低HASMCs的AAT活性,进而抑制其内源性SO2的生成。通过酶动力学研究,发现NO使AAT1纯化蛋白的Vmax值明显降低,Km值明显升高,说明NO直接抑制纯化蛋白AAT1活性,影响AAT1酶动力学特征。进一步探索NO抑制内源性SO2分子机制的研究发现,NO促进AAT1蛋白的亚硝基化修饰;LC-MS/MS结果揭示NO对AAT1蛋白的亚硝基化修饰发生在Cys192位点;巯基还原剂DTT或C192S突变可阻断NO诱导的AAT1亚硝基化修饰及其对AAT1酶活性的抑制效应。以上结果说明NO可通过亚硝基化AAT1 Cys192位点,抑制其活性,进而抑制内源性SO2生成。
综上,本研究首次揭示在病理条件下,内源性SO2体系作为重要的内源性防御体系,可在内源性NO损伤时代偿性激活,修复血管稳态失衡。VEC来源的NO抑制VSMC来源的SO2生成是VEC-VSMC基于气体介质进行细胞间通讯的一种新的分子机制。
