 ,11College of Physics and Electronic Engineering,
,11College of Physics and Electronic Engineering, 2School of Mathematics and Physics,
Received:2020-03-11Revised:2020-05-25Accepted:2020-05-25Online:2020-08-27

Abstract
Keywords:
PDF (768KB)MetadataMetricsRelated articlesExportEndNote|Ris|BibtexFavorite
Cite this article
Xuan-Hong Gao(高轩鸿), Lei Zhang(张磊), Zhi-Hong Jiao(焦志宏), Guo-Li Wang(王国利), Song-Feng Zhao(赵松峰). Determination of structure parameters in strong-field ionization models of atoms. Communications in Theoretical Physics, 2020, 72(9): 095504- doi:10.1088/1572-9494/aba248
1. Introduction
It is well-known that field ionization of atoms in a static electric field is one of the basic issues in quantum mechanics, which was pioneered theoretically by Oppenheimer in 1928 [1]. Although the accurate ionization rate of atoms can be obtained by solving the three-dimensional time-dependent Schrödinger equation mostly based on the single-active-electron approximation (SAE-TDSE) [2, 3], analytical ionization models are still desirable especially for interpreting the experimental data. The correct analytical formula for ionization rate of hydrogen atom in the ground state was obtained by Landau in [4]. Recently the weak-field asymptotic theory was successfully developed to study static ionization rate of atoms analytically [5]. Based on the Keldysh theory [6], the Ammosov–Delone–Krainov (ADK) [7] was proposed and became one of the most popular tunneling ionization models in strong-field physics. The ADK model was successfully extended to study the tunneling ionization of molecules (so called MO-ADK) [8–12] and to over-the-barrier regime empirically [13, 14]. Many other improvements on the ADK model have also been achieved by taking into account the polarizability effect [15] and the higher-order terms in the Wentzel–Kramers–Brillouin approximation [16], respectively. The Perelomov–Popov–Terent’ev (PPT) [17, 18] is another most used ionization model based on the nonadiabatic picture, which was generalized to study ionization probability of molecules by lasers (so called MO-PPT) [19, 20]. It has been confirmed that the PPT model can work very well in a wide intensity range covering from the multiphoton to tunneling ionization regimes, while the ADK model seriously underestimates ionization rate of atoms in the multiphoton ionization region [21, 22].Recall that the ionization rate of atoms depends on the field strength and structure parameter Cl in the ADK model [13] and the PPT model [20]. It is crucial to systematically determine and document structure parameters of atoms and ions used in the PPT and ADK models. However, structure parameters of only five rare gases were initially determined in [8], where electronic wave functions of noble gas atoms were calculated by using the self-interaction free density-functional theory [23]. We noticed that structure parameters (or expansion coefficients) were also systematically extracted from electronic wave functions obtained from the Hartree–Fock (HF) theory [24]. Since wave functions calculated from the quantum chemistry packages such as GAUSSIAN [25] or GAMESS [26] decay too rapidly to extract accurate structure parameters in the asymptotic region. The asymptotic properties of molecular wave functions can be partially improved by using the polarization-consistent basis sets [27, 28]. For atoms and diatomic molecules, the X2DHF program [29] can give the wave functions with high quality asymptotic tail. In our previous works [9, 30–33], we proposed an efficient method to calculate molecular wave functions with correct asymptotic behavior using the density-functional theory (DFT) and systematically determined structure parameters used in the MO-ADK [8] and MO-PPT [20].
In this paper, we extend the DFT method to extract accurate atomic structure parameters from wave functions in the asymptotic region for the 25 atoms, 24 positive ions and 13 negative ions, respectively. In section
2. Theoretical method
The theory part is separated into three subsections. We first consider the three-dimensional SAE-TDSE method to accurately calculate ionization probabilities of atoms. We will then present how to numerically calculate atomic potential and obtain wave functions with correct asymptotic behavior by solving time-independent Schrödinger equation with B-spline functions. Finally we briefly review basic equations of the ADK and PPT models.2.1. Method of solving the three-dimensional time-dependent Schrödinger equation
Based on the SAE approximation and the length gauge, the TDSE of atoms or ions in the presence of a linearly polarized laser field can be written as2.2. The DFT method
2.2.1. Construction of one-electron potentials for atoms and ions
Due to the spherical symmetry of atoms or ions, potentials do not depend on the orbital quantum number l and can be expanded asIn the modified Leeuwen–Baerends (LBα) model [30, 37, 38], the partial exchange-correlation potential is written as
2.2.2. Creation of wave functions with correct asymptotic tail for atoms or ions
After atomic potentials are numerically calculated with the DFT method, the electronic wave function with high quality asymptotic behavior can be obtained by solving the time-independent Schrödinger equation.Once wave functions with the correct asymptotic behavior are available, accurate structure parameters can be extracted by fitting the calculated radial wave functions Rnl(r) to the following form in the asymptotic region
2.3. The PPT and ADK models
According to the PPT mode [20], the cycle-averaged ionization rate can be analytically given byOnce cycle-averaged ionization rates of atoms or ions are obtained, we can calculate total ionization probability at the end of laser pulse by
3. Results and discussion
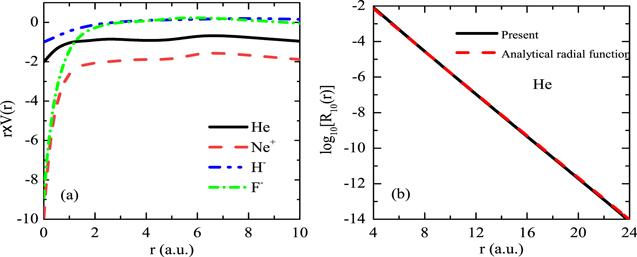
We first show the quality of potentials of atoms or ions in figure 1(a) and how to extract structure parameter Cl using the fitting procedure in figure 1(b). In figure 1(a), we can see the effective charge changes gradually from the nuclear charge to the asymptotic one. In the LBα model [30, 37, 38], we take β=0.01 and the adjustable parameter α is optimized to obtain the accurate ionization potential (IP) by comparing with the experimental IP from the NIST database [43]. The optimized α and comparison between the present calculated IPs and the experimental values are listed in table 1. In figure 1(b), it is clear that our calculated radial function decays exponentially and has a very good fitting to the asymptotic analytical form in equation (Figure 1.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 1.(a) The effective charge of He, Ne+, H−, and F−, where potentials are calculated by using the density-functional theory; (b) the structure parameter Cl of He is extracted from the electronic wave function in the asymptotic region by the fitting method.
Figure 2.
 New window|Download| PPT slide
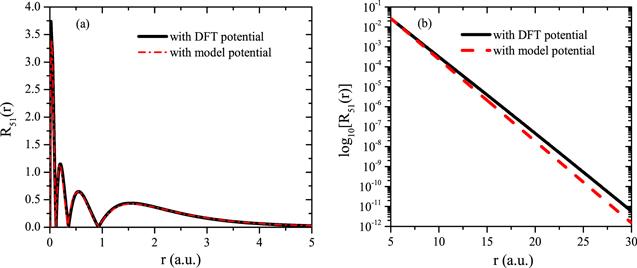
New window|Download| PPT slideFigure 2.Radial functions of Xe in the (a) small-r region; (b) large-r region.
Table 1.
Table 1.Direct comparison of the calculated IPs using the LBα model and the experimental values from the NIST database [43]. The optimized α parameters used in LBα model are also listed.
| Atom/ion | LBα (eV) | Ip (eV) | α | Atom/ion | LBα (eV) | Ip (eV) | α |
|---|---|---|---|---|---|---|---|
| H(1s) | 13.60 | 13.60 | — | He(1s) | 24.59 | 24.59 | 1.34 |
| Li(2s) | 5.41 | 5.39 | 1.24 | Be(2s) | 9.33 | 9.32 | 1.26 |
| B(2p) | 8.27 | 8.30 | 1.20 | C(2p) | 11.23 | 11.26 | 1.21 |
| N(2p) | 14.47 | 14.53 | 1.22 | O(2p) | 13.58 | 13.62 | 1.03 |
| F(2p) | 17.38 | 17.42 | 1.11 | Ne(2p) | 21.65 | 21.56 | 1.20 |
| Na(3s) | 5.12 | 5.14 | 0.87 | Mg(3s) | 7.78 | 7.65 | 0.75 |
| Al(3p) | 5.96 | 5.99 | 1.19 | Si(3p) | 8.16 | 8.15 | 1.20 |
| P(3p) | 10.44 | 10.49 | 1.22 | S(3p) | 10.36 | 10.36 | 1.07 |
| Cl(3p) | 12.84 | 12.97 | 1.13 | Ar(3p) | 15.75 | 15.76 | 1.22 |
| K(4s) | 4.35 | 4.34 | 0.54 | Ca(4s) | 6.12 | 6.11 | 0.41 |
| Br(4p) | 11.83 | 11.81 | 0.98 | Kr(4p) | 13.98 | 14.00 | 0.99 |
| Rb(5s) | 4.13 | 4.18 | 0.50 | I(5p) | 10.55 | 10.45 | 0.85 |
| Xe(5p) | 12.16 | 12.13 | 0.95 | He+(1s) | 54.48 | 54.42 | — |
| Li+(1s) | 75.67 | 75.64 | 1.31 | Be+(2s) | 18.24 | 18.21 | 1.22 |
| B+(2s) | 25.18 | 25.16 | 1.21 | C+(2p) | 24.37 | 24.38 | 1.25 |
| N+(2p) | 29.44 | 29.60 | 1.24 | O+(2p) | 35.23 | 35.12 | 1.26 |
| F+(2p) | 35.03 | 34.97 | 1.21 | Ne+(2p) | 40.95 | 40.96 | 1.14 |
| Na+(2p) | 47.17 | 47.29 | 1.23 | Mg+(3s) | 15.06 | 15.04 | 0.72 |
| Al+(3s) | 18.82 | 18.83 | 0.56 | Si+(3p) | 16.36 | 16.35 | 1.23 |
| P+(3p) | 19.74 | 19.77 | 1.22 | S+(3p) | 23.35 | 23.34 | 1.25 |
| Cl+(3p) | 23.75 | 23.81 | 1.09 | Ar+(3p) | 27.94 | 27.63 | 1.19 |
| K+(3p) | 31.63 | 31.63 | 1.20 | Ca+(4s) | 11.86 | 11.87 | 0.39 |
| Br+(4p) | 21.60 | 21.59 | 1.08 | Kr+(4p) | 24.30 | 24.36 | 0.95 |
| Rb+(4p) | 27.35 | 27.29 | 1.15 | I+(5p) | 19.10 | 19.13 | 1.04 |
| Xe+(5p) | 21.15 | 20.98 | 0.92 | H−(1s) | 0.76 | 0.75 | 1.17 |
| Li−(2s) | 0.62 | 0.62 | 1.13 | C−(2p) | 1.26 | 1.26 | 1.15 |
| O−(2p) | 1.46 | 1.46 | 1.05 | F−(2p) | 3.40 | 3.40 | 1.14 |
| Na−(3s) | 0.54 | 0.55 | 0.72 | Al−(3p) | 0.44 | 0.44 | 1.11 |
| Si−(3p) | 1.39 | 1.39 | 1.17 | P−(3p) | 0.75 | 0.75 | 1.01 |
| Cl−(3p) | 3.61 | 3.62 | 1.17 | K−(4s) | 0.50 | 0.50 | 0.48 |
| Br−(4p) | 3.38 | 3.37 | 1.00 | Rb−(5s) | 0.49 | 0.49 | 0.43 |
New window|CSV
Table 2.
Table 2.The fitted Cl structure parameters versus values from earlier [7, 8, 24, 44].
| Atom/ion | Cl | Atom/ion | Cl | Atom/ion | Cl | |||
|---|---|---|---|---|---|---|---|---|
| H(1s) | 2.00 | He(1s) | 2.42 | Ne(2p) | 1.47 | |||
| 2.00 | [7] | 2.98 | [7] | 2.77 | [7] | |||
| 3.13 | [8] | 2.10 | [8] | |||||
| 2.87 | [24] | 1.75 | [24] | |||||
| 2.96 | 1.95 | |||||||
| Ar(3p) | 1.85 | Kr(4p) | 2.17 | Xe(5p) | 4.39 | |||
| 2.25 | [7] | 2.05 | [7] | 1.80 | [7] | |||
| 2.44 | [8] | 2.49 | [8] | 2.57 | [8] | |||
| 2.11 | [24] | 2.22 | [24] | 2.40 | [24] | |||
| 2.51 | [44] | 2.59 | [44] | 2.72 | [44] | |||
| 2.44 | 2.01 | 2.55 | ||||||
| Li(2s) | 0.56 | Be(2s) | 1.44 | B(2p) | 0.66 | |||
| 0.59 | [7] | 1.36 | [7] | 1.17 | [7] | |||
| 0.82 | [24] | 1.62 | [24] | 0.88 | [24] | |||
| C(2p) | 0.92 | N(2p) | 1.21 | O(2p) | 0.95 | |||
| 1.67 | [7] | 2.11 | [7] | 2.00 | [7] | |||
| 1.30 | [24] | 1.50 | [24] | 1.30 | [24] | |||
| F(2p) | 1.19 | Na(3s) | 0.32 | Mg(3s) | 0.79 | |||
| 2.42 | [7] | 0.54 | [7] | 1.05 | [7] | |||
| 1.59 | [24] | 0.74 | [24] | 1.32 | [24] | |||
| Al(3p) | 0.46 | Si(3p) | 0.98 | P(3p) | 1.48 | |||
| 0.71 | [7] | 1.15 | [7] | 1.55 | [7] | |||
| 0.61 | [24] | 1.10 | [24] | 1.65 | [24] | |||
| S(3p) | 1.16 | Cl(3p) | 1.53 | K(4s) | 0.26 | |||
| 1.53 | [7] | 1.92 | [7] | 0.38 | [7] | |||
| 1.11 | [24] | 1.78 | [24] | 0.52 | [24] | |||
| Ca(4s) | 0.61 | Br(4p) | 1.71 | Rb(5s) | 0.31 | |||
| 0.74 | [7] | 1.76 | [7] | 0.34 | [7] | |||
| 0.95 | [24] | 1.83 | [24] | 0.48 | [24] | |||
| I(5p) | 3.72 | He+(1s) | 5.66 | Li+(1s) | 6.03 | |||
| 1.55 | [7] | 5.66 | [7] | 6.52 | [7] | |||
| 1.94 | [24] | 6.50 | [24] | |||||
| Be+(2s) | 2.21 | B+(2s) | 3.71 | C+(2p) | 2.17 | |||
| 1.96 | [7] | 3.04 | [7] | 2.93 | [7] | |||
| 2.67 | [24] | 6.70 | [24] | 2.53 | [24] | |||
| N+(2p) | 2.43 | O+(2p) | 2.88 | F+(2p) | 2.40 | |||
| 3.62 | [7] | 4.23 | [7] | 4.21 | [7] | |||
| 2.90 | [24] | 3.30 | [24] | 3.10 | [24] | |||
| Ne+(2p) | 2.67 | Na+(2p) | 2.74 | Mg+(3s) | 1.33 | |||
| 4.76 | [7] | 5.23 | [7] | 1.41 | [7] | |||
| 3.40 | [24] | 3.70 | [24] | 2.31 | [24] | |||
| 3.43 | ||||||||
| Al+(3s) | 2.14 | Si+(3p) | 1.58 | P+(3p) | 2.55 | |||
| 2.06 | [7] | 1.64 | [7] | 2.22 | [7] | |||
| 3.10 | [24] | 1.80 | [24] | 2.50 | [24] | |||
| S+(3p) | 2.86 | Cl+(3p) | 2.96 | Ar+(3p) | 3.47 | |||
| 2.78 | [7] | 2.85 | [7] | 3.37 | [7] | |||
| 3.20 | [24] | 3.10 | [24] | 3.40 | [24] | |||
| 3.81 | ||||||||
| K+(3p) | 3.59 | Ca+(4s) | 0.79 | Br+(4p) | 2.74 | |||
| 3.86 | [7] | 0.86 | [7] | 2.51 | [7] | |||
| 3.90 | [24] | 1.62 | [24] | 2.50 | [24] | |||
| Kr+(4p) | 3.55 | Rb+(4p) | 3.58 | I+(5p) | 3.60 | |||
| 2.93 | [7] | 3.33 | [7] | 2.11 | [7] | |||
| 3.70 | [24] | 3.82 | [24] | 2.90 | [24] | |||
| Xe+(5p) | 4.97 | H−(1s) | 0.47 | Li−(2s) | 0.78 | |||
| 2.42 | [7] | 1.12 | 1.00 | [24] | ||||
| 3.20 | [24] | |||||||
| C−(2p) | 0.45 | O−(2p) | 0.38 | F−(2p) | 0.65 | |||
| 0.74 | [24] | 0.65 | [24] | 0.84 | [24] | |||
| 0.76 | ||||||||
| Na−(3s) | 0.42 | Al−(3p) | 0.57 | Si−(3p) | 1.02 | |||
| 1.00 | [24] | 0.51 | [24] | 1.10 | [24] | |||
| P−(3p) | 0.55 | Cl−(3p) | 1.20 | K−(4s) | 0.57 | |||
| 0.90 | [24] | 1.34 | [24] | 0.90 | [24] | |||
| Br−(4p) | 1.41 | Rb−(5s) | 0.78 | |||||
| 1.49 | [24] | 0.80 | [24] |
New window|CSV
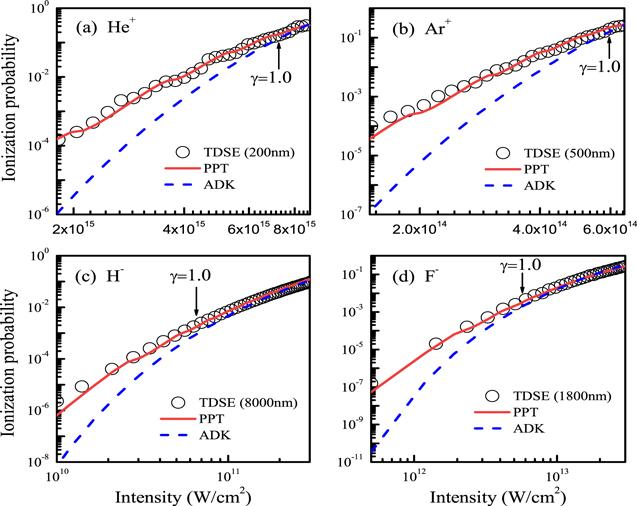
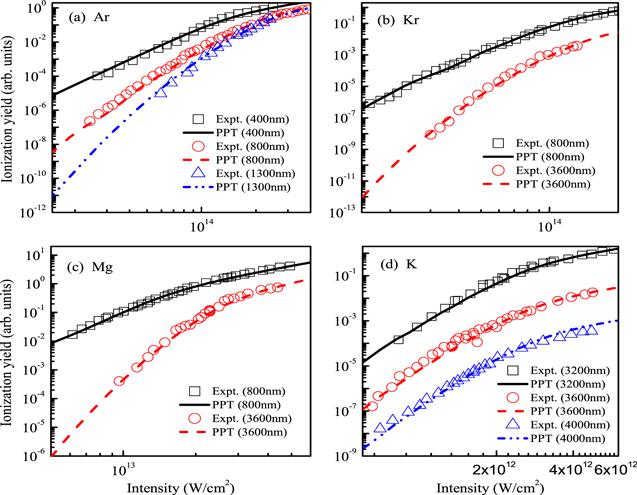
Let us turn to examine carefully the accuracy of these structure parameters in table 2. In figure 3, we compare ionization probabilities of He+, Ar+, H− and F− calculated using the PPT and ADK models with those obtained from the three-dimensional TDSE simulations. We mention that structure parameters extracted from our DFT wave functions are used in the PPT and ADK models. All the ionization probabilities from the PPT and ADK models are normalized to the TDSE results. One can see that all the ionization probabilities from the PPT model are in good agreement with the TDSE results in a wide intensity range covering from the multiphoton to tunneling ionization regimes, while the ADK model seriously underestimate ionization probabilities about 2–4 order of magnitude in the multiphoton region. Figure 4 compares ionization yields of Ar, Kr, Mg and K calculated from the PPT model (see equation (
Figure 3.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 3.Comparison of ionization probabilities obtained from three different methods: (a) He+, (b) Ar+, (c) H− and (d) F−. We take the laser field to be a Gaussian pulse with pulse duration (full width at half maximum, FWHM) of 15 fs.
Figure 4.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 4.Comparison of ionization yields at several different laser wavelengths: (a) Ar, (b) Kr, (c) Mg and (d) K. The volume effect of a focused laser pulse with FWHM of 90 fs is considered. The Experimental data are from [45].
4. Conclusions
In this paper, we systematically determined structure parameters used in the ADK and PPT models for 25 atoms, 24 positive ions and 13 negative ions based on the DFT. These structure parameters are very useful for calculating ionization rate using the ADK or PPT model. Especially we present structure parameters of 24 positive ions and of 13 negative ions which should be needed to study the sequential double ionization (or detachment) probabilities based on the ADK and PPT models. We mention that structure parameters for any highly charged ions can be extracted if necessary in the future. The reliability of these structure parameters are examined carefully by comparing ionization probabilities calculated by the ADK and PPT models with those three-dimensional TDSE results. By considering the volume effect, we found that all the calculated ionization yields using the PPT model can fit perfectly the experimental data at several different laser wavelengths both in multiphoton and tunneling ionization regimes.Acknowledgments
Project supported by the National Natural Science Foundation of China under Grant Nos. 11664035, 11864037 and 11765018. and the Foundation of Northwest Normal University (No. NWNU−LKQN−17−1).Reference By original order
By published year
By cited within times
By Impact factor
DOI:10.1103/PhysRev.31.66 [Cited within: 1]
DOI:10.1103/PhysRevA.35.445 [Cited within: 1]
DOI:10.1016/S0301-0104(97)00063-3 [Cited within: 2]
[Cited within: 1]
DOI:10.1103/PhysRevA.84.053423 [Cited within: 1]
[Cited within: 1]
DOI:10.1007/978-3-642-85691-4_3 [Cited within: 52]
DOI:10.1103/PhysRevA.66.033402 [Cited within: 10]
DOI:10.1103/PhysRevA.81.033423 [Cited within: 1]
DOI:10.1088/0953-4075/44/3/035601
DOI:10.1103/PhysRevA.71.013418
DOI:10.1103/PhysRevA.76.043419 [Cited within: 1]
DOI:10.1088/0953-4075/38/15/001 [Cited within: 7]
DOI:10.1103/PhysRevA.90.043410 [Cited within: 1]
DOI:10.1103/PhysRevLett.95.073001 [Cited within: 1]
DOI:10.1103/PhysRevA.87.045403 [Cited within: 1]
[Cited within: 2]
DOI:10.1088/0953-4075/25/19/011 [Cited within: 2]
DOI:10.1103/PhysRevA.70.025401 [Cited within: 1]
DOI:10.1103/PhysRevA.93.023413 [Cited within: 4]
DOI:10.1088/1674-1056/21/11/113101 [Cited within: 2]
DOI:10.1016/j.optcom.2013.09.074 [Cited within: 1]
DOI:10.1103/PhysRevA.55.3406 [Cited within: 1]
[Cited within: 62]
[Cited within: 2]
DOI:10.1002/jcc.540141112 [Cited within: 1]
DOI:10.1103/PhysRevA.87.013406 [Cited within: 1]
DOI:10.1103/PhysRevA.89.033412 [Cited within: 1]
DOI:10.1016/0010-4655(96)00098-7 [Cited within: 2]
DOI:10.1103/PhysRevA.82.035402 [Cited within: 4]
DOI:10.1080/00268976.2013.833349
DOI:10.1088/0253-6102/65/3/366
DOI:10.1088/0253-6102/67/3/289 [Cited within: 1]
DOI:10.1103/PhysRevA.77.053410 [Cited within: 2]
DOI:10.1103/PhysRevA.48.4654 [Cited within: 2]
DOI:10.1103/PhysRevA.78.063404 [Cited within: 1]
DOI:10.1063/1.480688 [Cited within: 2]
DOI:10.1063/1.1904587 [Cited within: 2]
DOI:10.1088/0034-4885/64/12/205 [Cited within: 1]
DOI:10.1002/qua.24582
DOI:10.1088/0953-4075/37/20/005 [Cited within: 1]
DOI:10.1364/JOSAB.8.000858 [Cited within: 1]
[Cited within: 2]
DOI:10.1103/PhysRevA.71.023411 [Cited within: 5]
DOI:10.1103/PhysRevA.96.063417 [Cited within: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}