1.BeiJing JiaoTong University HaiBin College, Huanghua 061199, China 2.Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China
Fund Project:Project supported by the Emergency Management Program of the National Natural Science Foundation of China(Special Funds for Theoretical Physics)(Grant No. 11547224) and the Research and Practice of the Higher Education reform in Hebei Province, China (Grant No. 2018GJJG649).
Received Date:24 January 2019
Accepted Date:26 April 2019
Available Online:01 July 2019
Published Online:05 July 2019
Abstract:One of the key issues for scale applications of hydrogen energy is the availability of safe, efficient and ecnomicical hydrogen storage technologies. In the past few years, light metal hydrides have attracted considerable attention due to their high hydrogen capacity. With a hydrogen capacity up to ~6.5 wt%, Li2NH is regarded as one of the most promising hydrogen storage materials. Although the hydrogen physical and thermodynamic properties of Li2NH have been studied, the electronic structure, phonon vibration mode and thermodynamic properties of Li2NH have not yet been resolved. In this paper, by using the first principles based on the density functional theory (DFT), we investigate the electronic structure, lattice dynamical and thermodynamic properties of Li2NH in detail.Firstly, the structure of Li2NH is optimized and the lattice parameters and total energy of the crystals are calculated. As shown by the calculation results, the lattice parameters are in good agreement with previous theoretical and experimental results. Our lowest-energy structure of Li2NH has orthorhombic Pnma symmetry at T=0 K for all of the proposed structures. Secondly, the electronic band-structure studies reveal that Li2NH has a small band gap of about 2.0 eV. The analysis of total and partial density of states of Li2NH show that the bonding between the N and H has a covalent character. Thirdly, the lattice dynamical properties of Li2NH are investgated at the corresponding equilibrium states. These results show that only the phonon dispersion curves of Li2NH (Pnma) without negative frequencies are calculated along the high-symmetry points. The optical modes of phonon frequencies at Γ point are assigned as Raman and Infrared-active modes. Based on the calculated phonon density of states, the thermodynamic properties are computed, such as the Helmholtz free energy, internal energy, entropy and the constant-volume specific heat versus temperature. The calculation results may explore the applications in areas of hydrogen storage for Li-N-H, which is of great importance forusing hydrogen in the future. Keywords:first-principles/ Li-N-H hydrogen storage/ lattice dynamics/ thermodynamic properties
a = 5.159[17],5.223[17],5.074[17],5.649[16],5.1076[18],5.2968[9]
–1920.4448
$Pnma$
a = 7.781 b = 3.623 c = 4.902
a = 7.733[8],7.704[16],7.742[17],7.753[17],7.775[17] b = 3.60[8],3.75[16],3.604[17],3.609[17],3.618[17] c = 4.872[8],5.074[16],4.883[17],4.890[17],4.881[17]
表2Li2NH($Pnma$)在布里渊区中心Γ点的光学模声子频率(单位:cm–1) Table2.Phonon frequencies (unit:cm–1) at the Γ point of Li2NH.
23.4.热力学性质 -->
3.4.热力学性质
了解材料的热力学性质对于研究其稳定性和化学反应是至关重要的. 基于图7得到的声子态密度, 按照晶格振动理论很容易得到Li2NH的热力学性质. 在简谐近似内, 晶格振动对Helmholtz自由能ΔF的贡献为[32] 图 7 Li2NH($Pnma$)的声子态密度 Figure7. The phonon density of states of Li2NH ($Pnma$).
$\Delta F=3 n N k_{B} T \int_{0}^{\omega_{\max }} \ln \left\{2 \sin \frac{\hbar \omega}{2 k_{B} T}\right\} g(\omega) d \omega, $






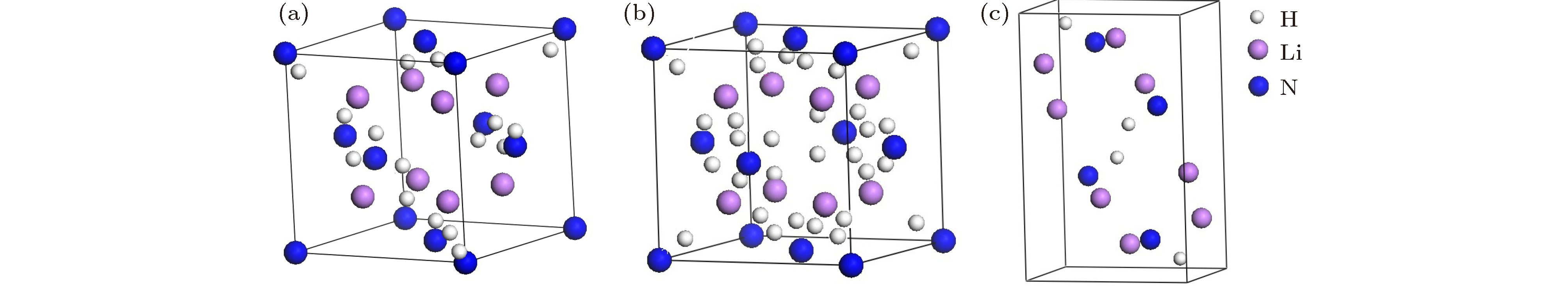 图 1 Li2NH的三种晶体结构图 (a) Li2NH (
图 1 Li2NH的三种晶体结构图 (a) Li2NH (
















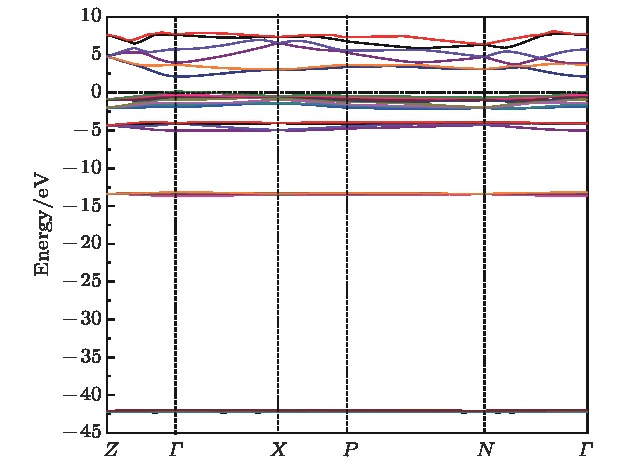 图 2 Li2NH的能带结构
图 2 Li2NH的能带结构 图 3 Li2NH的总态密度和分波态密度
图 3 Li2NH的总态密度和分波态密度



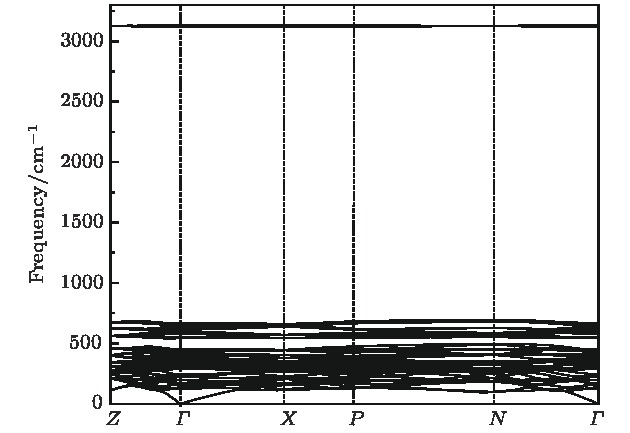 图 4 Li2NH(
图 4 Li2NH(

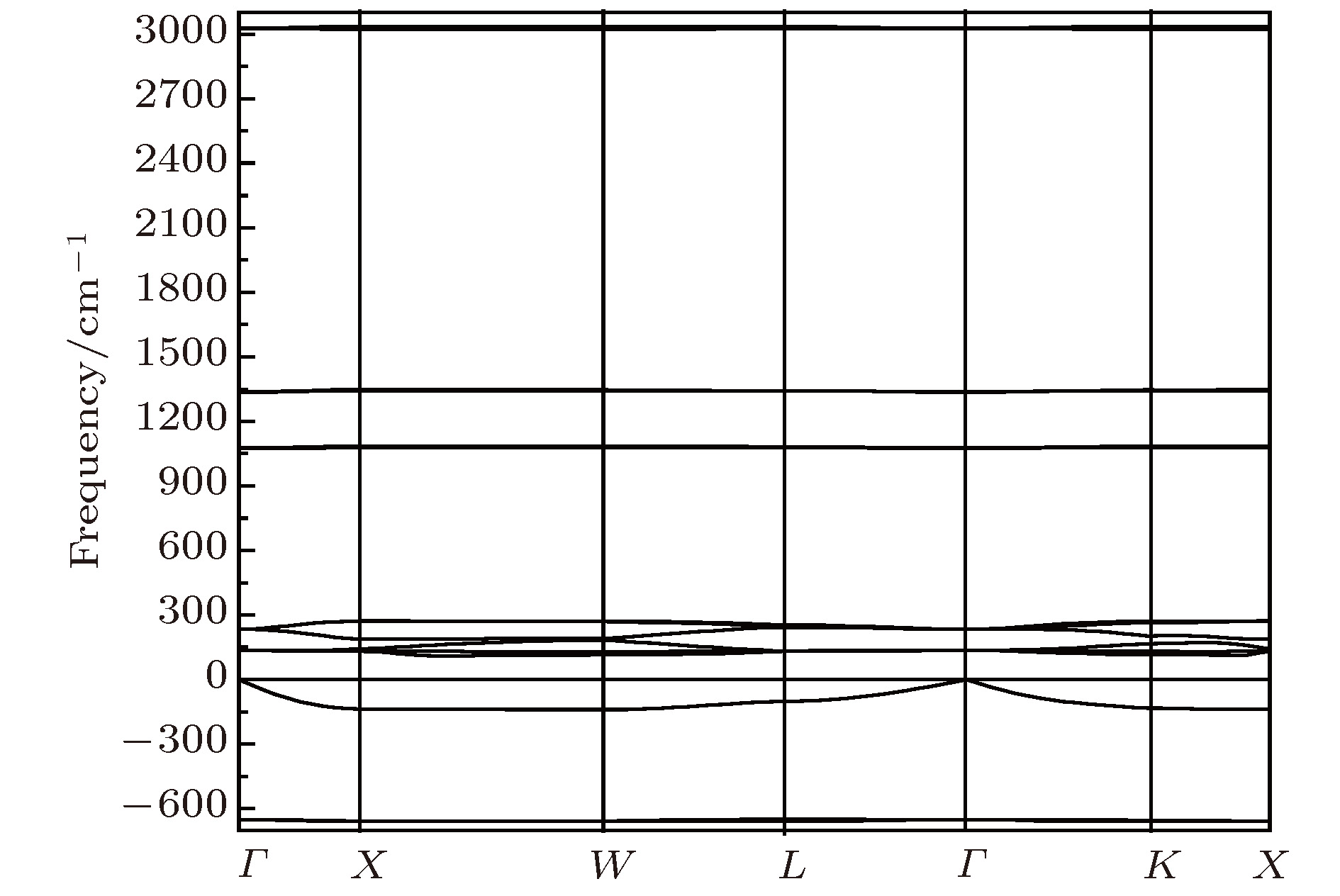 图 5 Li2NH(
图 5 Li2NH(

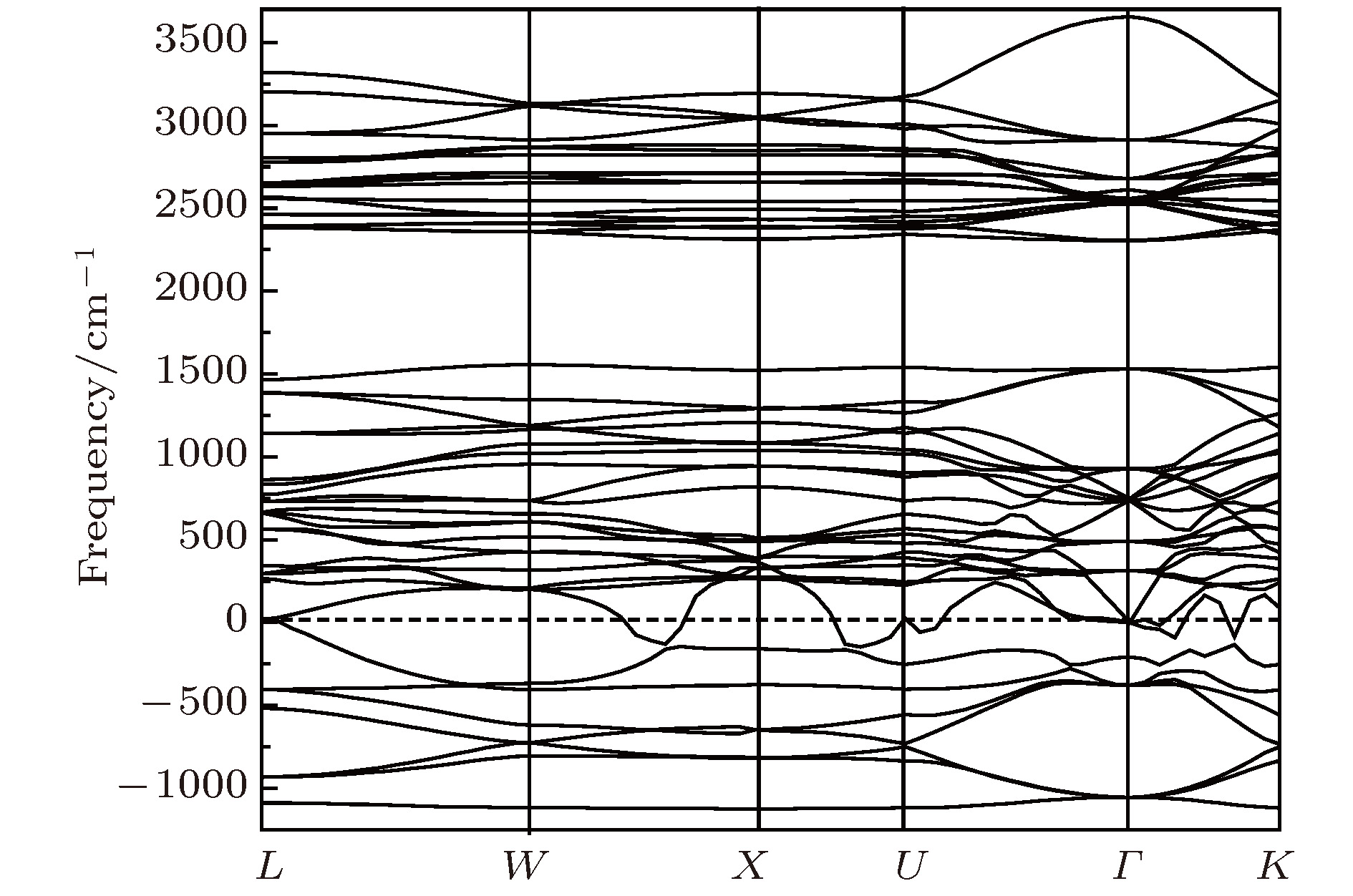 图 6 Li2NH(
图 6 Li2NH(





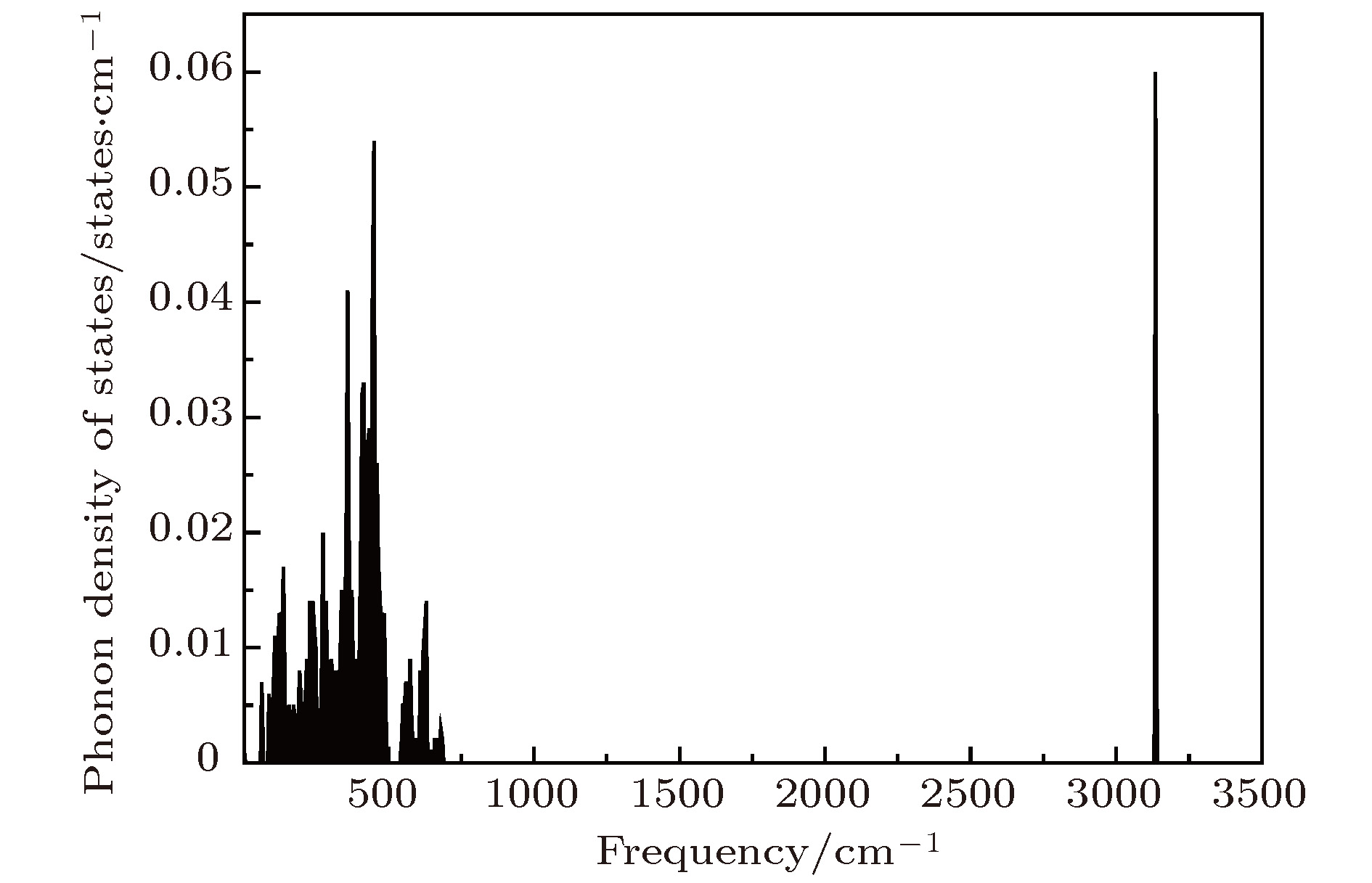 图 7 Li2NH(
图 7 Li2NH(






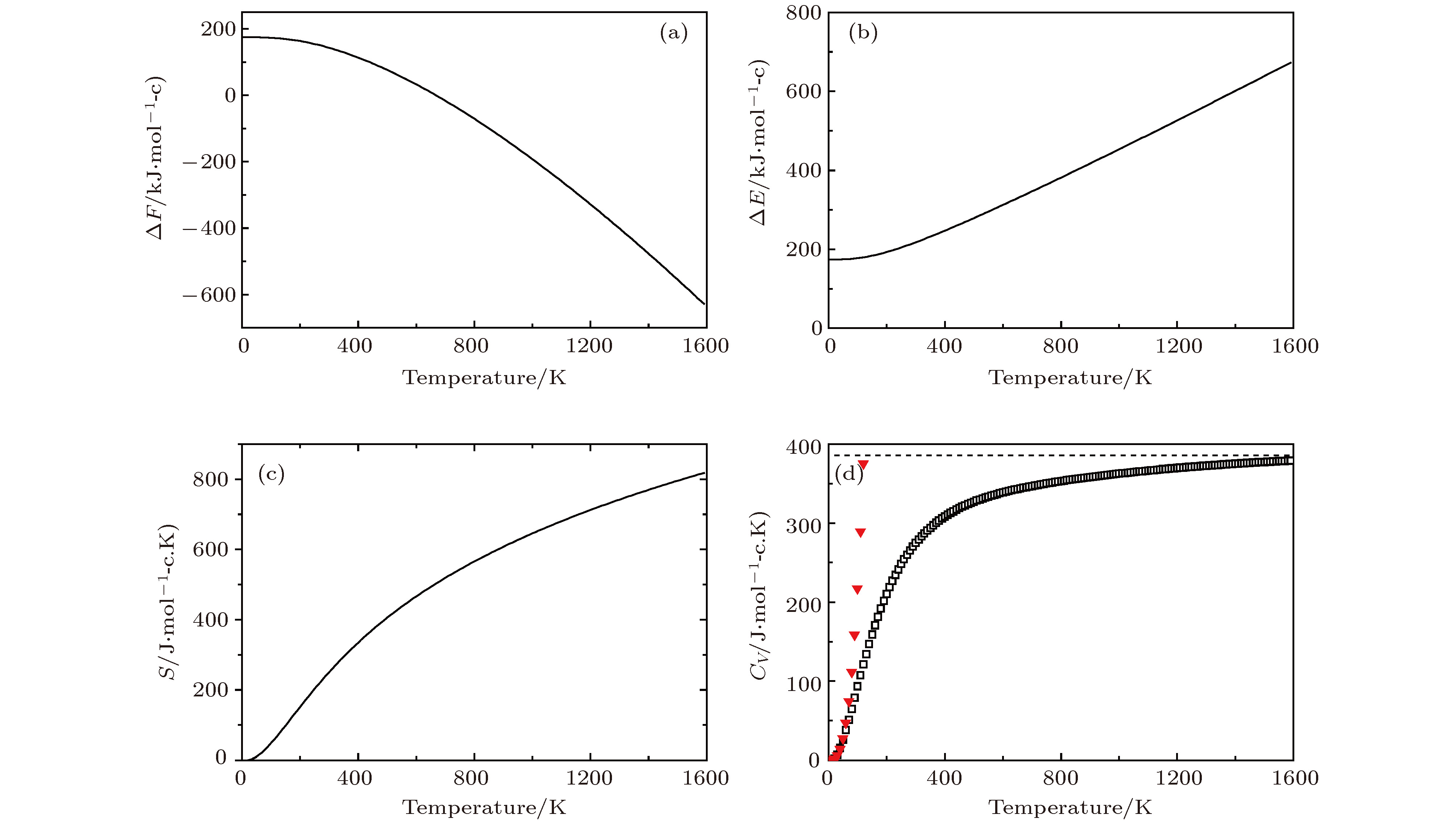 图 8 Li2NH的热力学性质 (a)晶格振动对自由能的贡献ΔF; (b)内能ΔE随温度的变化; (c)熵S随温度的变化; (d)热容Cv随温度的变化 (▼表示Debye的T3定律)
图 8 Li2NH的热力学性质 (a)晶格振动对自由能的贡献ΔF; (b)内能ΔE随温度的变化; (c)熵S随温度的变化; (d)热容Cv随温度的变化 (▼表示Debye的T3定律)

