全文HTML
--> --> -->稀土元素具有大的离子半径, 高的电子电荷, 以及强的自极化能力, 作为掺杂元素, 不仅对于尖晶石型钴铁氧体的电子结构和磁性有明显的调节作用[4], 而且在锂离子电池正极材料的改性上起到了积极的作用. 通过稀土掺杂的方式可以有效改善正极材料的性能. 近些年, 实验方面对于稀土元素掺杂锂离子电池正极材料的研究有很多, 比如: LiCoO2[5—7], LiMn2O4[8—12], LiFePO4[13,14]和LiNi1/3Co1/3Mn1/3O2[15]等. Yang等[16]报道了LiMn2O4电池正极材料的稀土掺杂, 掺杂后形成的LiMn2–xRExO4(x = 0.01, RE = Y, Nd, Gd, Ce) 体系晶格参数由于掺杂稀土离子而增大, 形成更为稳定的骨架结构, 显著提高了电极材料的循环性能. 此外, 采用大离子半径的稀土元素进行掺杂, 从某种程度上扩展了正极材料中的三维扩散通道, 促进了Li离子的迁移. Sun等[17]研究了LiMn2–xRExO4 (0 ≤ x ≤ 0.01, RE = La, Ce, Nd, Sm) 的正极材料. 研究结果表明, 与纯的LiMn2O4相比, 稀土掺杂后的材料循环性能和倍率性能均有显著提高. 不仅是LiMn2O4正极材料, Zhang等[18]还在LiFePO4中用La替代Li实现了可逆容量、电导率和Li离子迁移率的显著提高. 在三元材料的掺杂研究方面, 实验表明Li[Ni1/3Co1/3Mn1/3]1–xRexO2 (0 ≤ x ≤ 0.04, Re = La, Ce, Pr) 具有较高放电能力和较好的循环性能, 其原因是稀土元素的掺入成功抑制了循环过程中的电荷转移[19].
层状的富锂锰基固溶体因其具有高容量、高电压、成本低以及环保等优点, 是具有潜力的下一代锂离子动力电池正极材料. 富锂锰基正极在不断的充放电过程中存在的循环性能差、电压衰减、向尖晶石结构的不可逆转变、不可逆容量大等问题, 与富锂锰基材料中的主要成分Li2MnO3的活化过程密切相关[20]. 不仅如此, Li2MnO3在深度脱锂的过程中会发生释氧. 释氧对于电池的安全来说是一个不容忽视的隐患, 它会导致电池的鼓包甚至是爆炸[21]. 为了提高Li2MnO3的稳定性和电化学性质, 人们提出了掺杂改性的方案. Gao等[22]通过第一性原理计算研究发现Mn位Mo掺杂能够减小带隙, 促进Li离子输运以及起到抑制释氧的作用. 此外, 我们以前的理论研究表明, Li2MnO3 利用P掺杂可以抑制氧缺陷, 阻止氧气的形成, 并且通过抑制由层状材料到尖晶石结构的相变, 从而提高结构的稳定性[23].
上述关于Li2MnO3的研究表明, 掺杂可以有效改善电池正极材料的性能. 尽管Li2MnO3的掺杂研究很多, 但有关稀土掺杂的研究工作尚未见报道. 由于稀土掺杂在锂离子电池电极材料的改性中能起到良好的效果, 并且采用理论模拟的方法能够方便地预测材料的性能[24], 所以在本研究工作中, 采用第一性原理计算的方法研究稀土元素(La, Ce, Pr, Sm)掺杂Li2MnO3的原子结构、电子结构以及离子迁移动力学性质, 从理论上预测稀土掺杂对Li2MnO3的改性效果.
3.1.稀土掺杂的Li2MnO3的原子结构
Li2MnO3是一种空间群为C2/m的富锂锰基正极材料. 其中, Li占据晶体的4h, 2c和2b位置, O占据了8j和4i位置, Mn占据了4g位置. 2b位置的Li离子在过渡金属层中, 4h和2c位置的Li离子在Li层中[35]. 我们对2 × 1 × 2超胞结构进行优化, 优化后的晶格参数列于表1中, 其中, 2a = 10.030 ?, b = 8.670 ?, 2c = 10.191 ?, 与实验值非常接近[34]. 随后, 对未掺杂Li2MnO3的完美结构进行稀土掺杂. 所有的掺杂都是将一个Mn原子替换成稀土原子(La, Ce, Pr, Sm), 如图1所示. 结构优化之后, 我们发现晶胞的形状并没有改变, 依然保持未掺杂时的结构, 但晶格常数明显增大, 同时晶胞的体积也增加, 如表1所列, 显然这是因为稀土元素的离子半径比Mn离子大所导致的. 此外, 对比四种稀土元素掺杂后的晶格体积可以发现, La掺杂的Li2MnO3体积变化最大, 而Sm掺杂的体系体积变化最小. 尽管实验上目前还缺少Li2MnO3稀土掺杂的结构数据, 但可以发现有关锂离子电池正极材料LiMn2O4的结果. 有趣的是, Sun等[17]以及Iqbal和Ahmad[9]的实验结果表明LiMn2O4进行稀土掺杂后, 晶格常数相比于未掺杂时的减少了. 他们给出的解释是由于稀土离子替位Mn离子后形成的RE—O键比Mn—O键更强, 因此RE—O键长比Mn—O键长更短, 从而导致了晶格常数的减小. 然而, 根据我们获得的Li2MnO3的晶格优化结果, 我们推断稀土离子替换Mn离子并不能减小稀土离子与O离子之间的键长, 所以实验上观测到的LiMn2O4由于稀土掺杂造成的晶格常数减小还存在其他的影响因素, 此方向值得进一步研究.| 体系 | 稀土元素的磁矩/μB | 2a/? | b/? | 2c/? | V/?3 |
| Li2MnO3(exp)[36] | — | 9.874 | 8.532 | 10.060 | — |
| Li2MnO3 | — | 10.030 | 8.670 | 10.191 | 835.566 |
| La-Li2MnO3 | 0 | 10.244 | 8.828 | 10.317 | 877.527 |
| Ce-Li2MnO3 | 0 | 10.157 | 8.769 | 10.266 | 861.058 |
| Pr-Li2MnO3 | 1 | 10.151 | 8.765 | 10.263 | 859.917 |
| Sm-Li2MnO3 | 0 | 10.136 | 8.755 | 10.248 | 856.630 |
表1未掺杂与稀土掺杂的Li2MnO3的晶格常数、超胞体积与稀土元素的磁矩
Table1.Lattice constants, volume of supercell, magnetic moment of rare-earth atom of Li2MnO3 without and with rare-earth doping
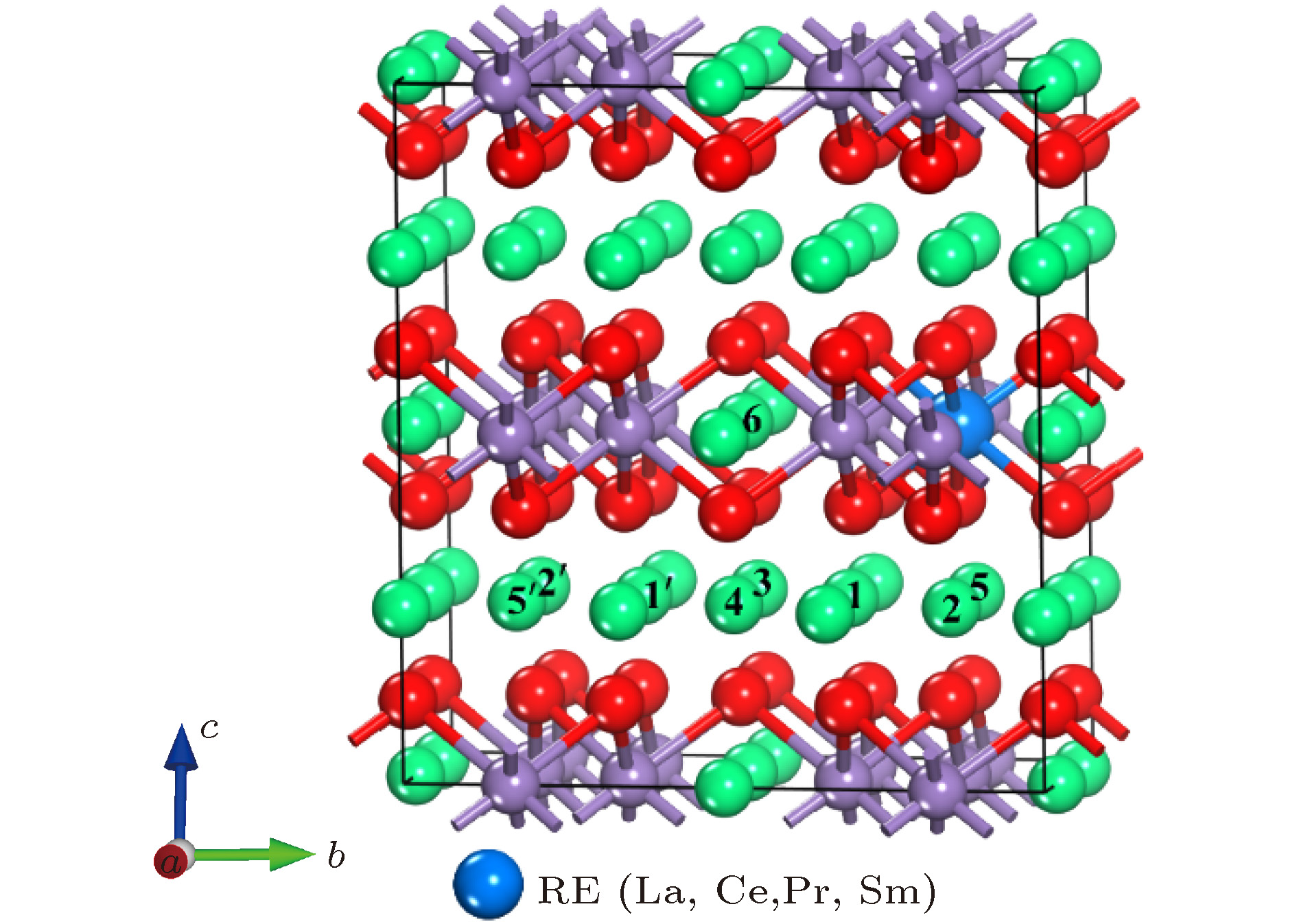 图 1 Li2MnO3中稀土掺杂位置与Li离子的9种不同等价位示意图
图 1 Li2MnO3中稀土掺杂位置与Li离子的9种不同等价位示意图Figure1. Crystal structure of Li2MnO3 with rare-earth doping sites and nine Li sites
为了了解稀土掺杂对Li2MnO3结构键长的影响, 表2列出了稀土掺杂的Li2MnO3优化之后的有关稀土离子与最近邻O离子之间的详细键长. 为了更好地进行比较, 表2还给出了未掺杂Li2MnO3中MnO6八面体的键长. 在未进行稀土掺杂时, MnO6八面体中的六条Mn—O键长几乎相等, 其中四条键长为1.941 ?, 另外两条分别1.934和1.949 ?. 当Mn离子被La离子替换后, La—O之间的键长明显扩大, 六条键长每两条相等, 三组键长分别为2.374, 2.385和2.401 ?. 受到La掺杂的影响, La近邻的MnO6八面体中的Mn—O键长也发生了变化, 键长范围在1.910—1.967 ?之间. 当Mn离子被Ce离子替代后, Ce—O键长分别为2.198, 2.215和2.218 ?, 其近邻的MnO6八面体中Mn—O键长范围在1.931— 1.955 ?之间. 当Pr和Sm离子替换Mn离子之后, Pr—O和Sm—O的键长分别在2.193—2.211 ?和2.170—2.183 ?范围内, 近邻MnO6八面体中Mn-—O键长变化范围分别为1.940—1.955 ?和1.916—1.964 ?. 计算数据表明, 当Li2MnO3中掺入稀土元素后, 稀土离子与O离子之间的键长与未掺杂结构中的Mn—O键长相比都显著增大, 从而扩大了整个晶体的晶格常数.
| Mn4+ | La3+ | Ce4+ | Pr4+ | Sm4+ | |
| dM—O (M = Mn, La, Ce, Pr, Sm)/? | 1.941 | 2.374 | 2.215 | 2.193 | 2.181 |
| 1.934 | 2.385 | 2.198 | 2.208 | 2.183 | |
| 1.949 | 2.401 | 2.218 | 2.211 | 2.170 |
表2未掺杂Li2MnO3中的Mn离子与最近邻O离子之间的键长和稀土掺杂结构中稀土离子与最近邻O离子之间的键长
Table2.Distance between Mn and the nearest neighboring O in undoped Li2MnO3 and distance between rare-earth ion and the nearest neighboring O in rare-earth-doped Li2MnO3
2
3.2.稀土掺杂的Li2MnO3的电子结构性质
图2是未掺杂Li2MnO3的电子态密度图. 从图2中可以看出, Li2MnO3是半导体材料, 计算带隙约为1.60 eV. 此外, 计算数据显示, Mn在体系中呈现+4价, 磁矩为3 μB. 根据晶体场理论, Mn4+离子中的3个电子占据Mn-3d轨道中的t2g轨道, eg轨道为全空的状态. 因为Mn4+离子很稳定, 电子不易失去, 所以在脱锂过程中, Mn的价态保持不变, O会补偿电荷, 从而使结构不稳定. 图3展示了La, Ce, Pr和Sm掺杂后的Li2MnO3的总态密度与稀土离子的投影态密度. 由图3(a)可以看出, 当掺入La时, Li2MnO3由半导体转变为金属性质, 有电子态穿过费米能级. 原因是由于La原子的价电子构型为5d16s2, 最外层有3个电子, 当掺入Li2MnO3后形成+3价的离子La3+. 而Li2MnO3中的Mn离子是+4价的离子. 因此在这种非等价离子替换的情况下, 部分O的电子态由原来的占据态转变为非占据态, 从而形成金属性的电子结构, 如图3(a)中的O原子的投影态密度所示. 与之不同的是, Ce和Pr掺杂后的Li2MnO3的电子态密度图与未掺杂情况下的非常相似, 都显示出半导体的特性, 如图3(b)和图3(c)所示. 从相似的电子态密度图上可以判断出, Ce和Pr掺杂之后都形成了+4价的离子态, 与Mn4+的价态相等, 是等价态离子替换, 从而没有改变本质的导电性. 为了进一步证实, 我们分析了掺杂后稀土离子的磁矩, 列于表1中. Ce最外层电子排布为4f1 5d1 6s2, 当形成+4价离子时, 外层电子全部失去, 导致磁矩为0. 类似地, Pr原子外层电子排布是4f3 6s2, 形成+4价离子后剩余一个f电子, 总磁矩为1 μB. 因此, 我们对价态的分析与磁矩计算结果是相符的. 尽管Ce和Pr掺杂的Li2MnO3表现为半导体特性, 但带隙较未掺杂的Li2MnO3变小了, 带隙分别降为1.30和1.40 eV.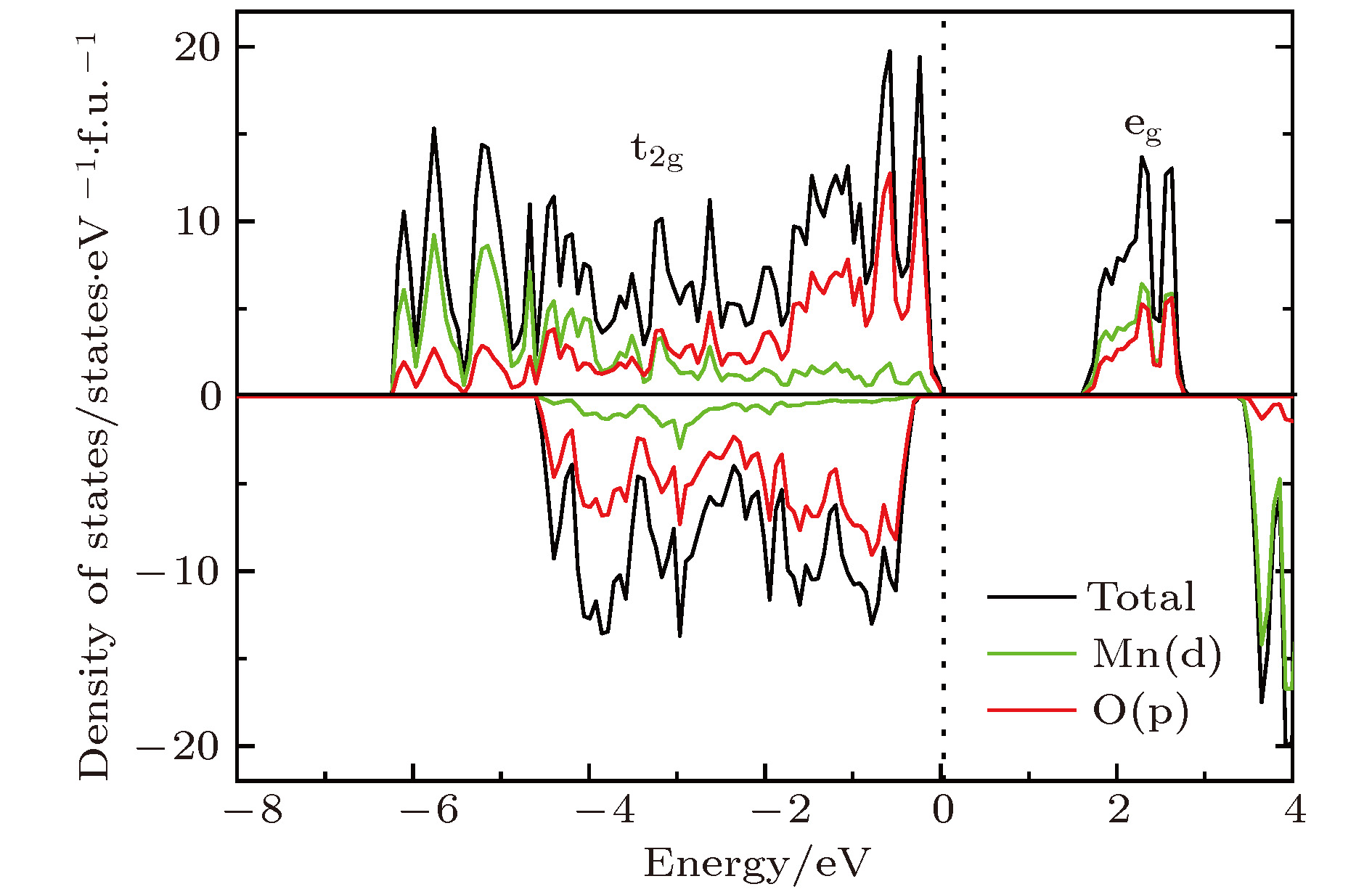 图 2 未掺杂Li2MnO3的电子态密度图
图 2 未掺杂Li2MnO3的电子态密度图Figure2. Density of states of Li2MnO3 without doping
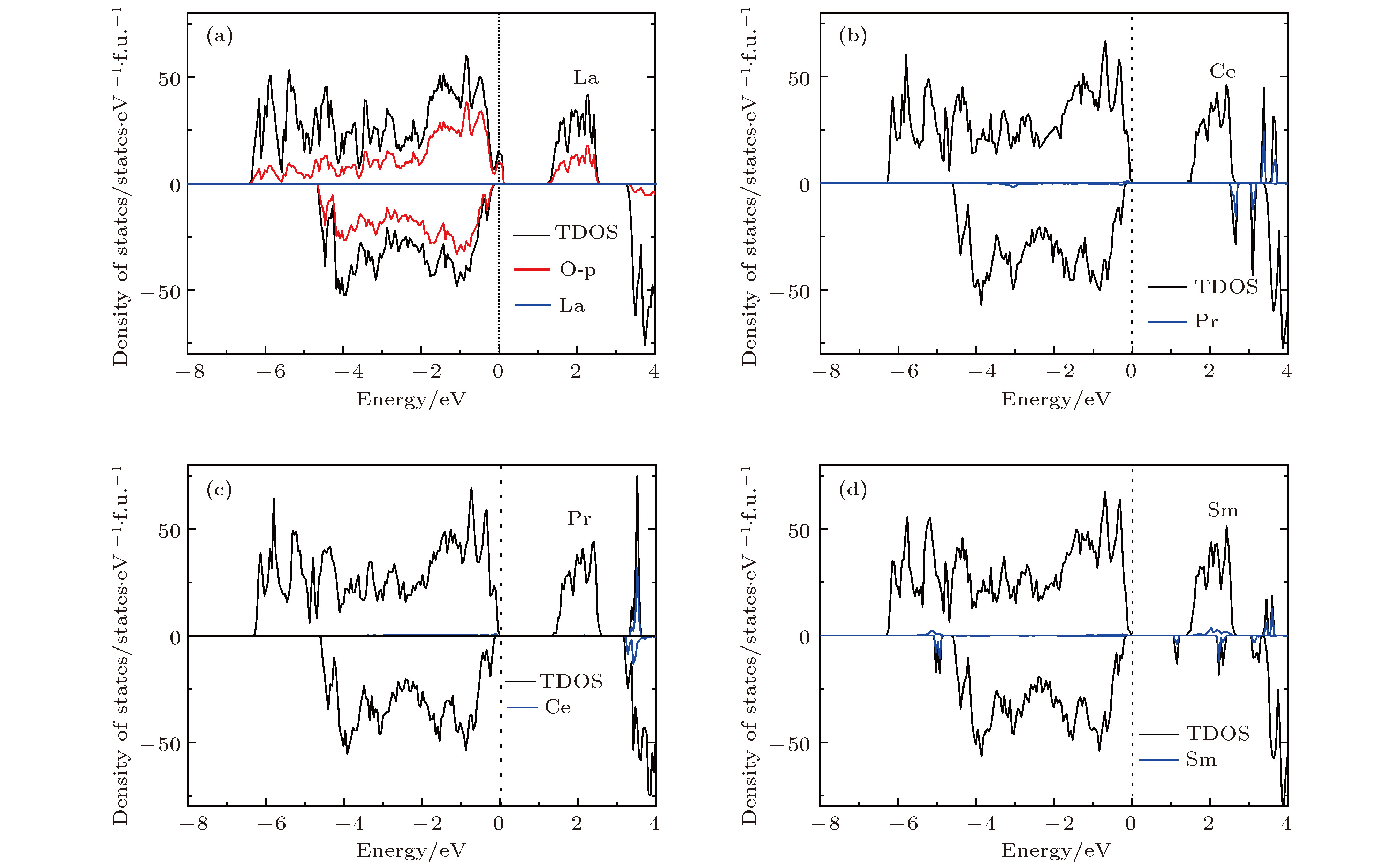 图 3 稀土元素 (a) La, (b) Ce, (c) Pr, (d) Sm掺杂的Li2MnO3的电子态密度
图 3 稀土元素 (a) La, (b) Ce, (c) Pr, (d) Sm掺杂的Li2MnO3的电子态密度Figure3. Density of states of Li2MnO3 with (a) La, (b) Ce, (c) Pr, and (d) Sm doping
图3(d)显示的是Sm掺杂的Li2MnO3的电子态密度. 显然, 该电子态密度依然保持了半导体的特性, 但带隙减少为0.91 eV. 从总的态密度图上可以看出, 带隙减小的主要原因是在费米能级附近出现了一个较为局域的带隙态. 为了进一步分析该带隙态, 我们给出了Sm的f轨道的分波态密度, 同样显示于图3(d)中. 分波态密度结果表明带隙态几乎是由Sm的f轨道贡献的. 虽然影响带隙减小的带隙态相对比较局域, 但在某种程度上对于体系的导电性起到增强的作用.
2
3.3.稀土掺杂的Li2MnO3中的Li离子迁移动力学
由于Li离子在电极化合物中的迁移率是体现可充电锂离子电池倍率性能的一个关键因素, 因此研究Li离子迁移的能垒对锂离子电池的应用至关重要. 在稀土掺杂的Li2MnO3中, 因为Ce, Pr和Sm都是相同的价态, 所以它们对于Li离子迁移的影响是类似的. 对比表1中Ce, Pr和Sm掺杂后的Li2MnO3的晶格常数以及表2中Ce—O, Pr—O和Sm—O键长可以发现, Ce, Pr和Sm掺杂产生的结构影响相差很小. 因此, 我们选取具有代表性的La和Ce元素掺杂的Li2MnO3体系, 研究其中的Li离子迁移动力学.参考Xiao等[37]报道的关于Li2MnO3中Li离子迁移路径的分析, 并且结合稀土元素的掺杂位置, 本文中我们定义了9个Li离子的位置, 示于图1中. 因为超胞的产生是原胞在a方向扩了一倍, 根据周期性边界条件, 以6号, 4号和3号Li原子为镜面, 整个晶胞左右对称. 因为La/Ce的掺入, 体系不再对称, 即使相同的wyckoff位置也具有不同的迁移类型. 为了便于描述, 相对于掺杂的稀土离子而言, 我们把靠近稀土离子一边的Li位定义为近邻位, 包括1, 2和5号位; 把远离稀土离子一边的Li位定义为远端位, 包括1', 2'和5'号位, 剩下的6, 3和4号位则作为中间位. 其中, 6是2b位, 3和4都是2c位, 1, 2, 5 (或1', 2', 5')都是4h位. 为了全面分析迁移路径, 我们总共考虑了11条迁移路径的能垒, 如表3所列, 并与未掺杂时的迁移路径进行比较.
| 未掺杂 | 稀土掺杂 | ||
| 2b-2c | 6-3 | 6-3 | |
| 2b-4h | 6-1 | 6-1 | 6-1' |
| 4h-2c | 1-3 | 1-3 | 1'-3 |
| 4h-2c | 1-4 | 1'-4 | |
| 4h-4h | 1-5 | 1-5 | 1'-5' |
| 4h-4h | 1-2 | 1'-2' | |
表3两种近邻结构中的Li离子迁移路径
Table3.Diffusion paths of Li ions in two different structures with neighboring Li ions
Li离子在La掺杂的Li2MnO3中的迁移情况分成两类, 如图4所示, 左边一列图4(a), 4(b), 4(d), 4(f), 4(h), 4(j)为近邻位的Li离子的迁移势垒, 右边一列图4(c), 4(e), 4(g), 4(i), 4(k)是远端位的Li离子的迁移势垒. 图4中远端位的离子迁移势垒范围为0.36—0.53 eV, 与未掺杂时相同wyckoff位的结果(0.51—0.84 eV)[37]相比, 有了明显降低. 这主要是因为Li离子迁移势垒的大小与过渡金属层之间的空间密切相关. 相比于未掺杂的Li2MnO3, La掺杂后c方向的晶格常数明显增加, 从而拉大了迁移空间, 减小了迁移势垒, 使得Li离子的迁移更容易发生. 与远端位的离子迁移情况不同, 近邻位的Li离子迁移的势垒变化范围非常大, 具体为0.01—1.03 eV. 我们推断如此大的势垒变化与La掺杂之后的局域结构变化大有关. 为了证实这一点, 我们分析了La离子周围的局域结构. 结果表明, La掺入后形成了La—O键, 其键长比Mn—O键长长, 会局域性地压缩Li层上下O与O之间的层间距. 我们的数据显示, 远端位的Li层上下O与O之间层间距大约为2.817 ?, 而稀土离子正下方的O与O层间距只有2.724 ?. 因此围绕着La周围的离子迁移, 势垒很大, 如6-1路径的0.87 eV和1-5路径的1.03 eV. 从上面的结果可以得出, 一方面, 稀土掺杂整体上可以降低Li离子的迁移势垒, 提高迁移速率; 另一方面, 稀土离子的掺入改变了局域结构, 使得周围的势能面起伏更大, 不利于Li离子的迁移. 所以, 稀土离子的掺杂浓度对于Li离子的迁移有着重要的影响, 需要进一步优化. 值得注意的是, 我们计算中只将1个Mn原子替换成稀土离子, 计算得到的掺杂比例为6.25%. 实验上, Sun等[17]对于LiMn2O4的掺杂比例小于1%, 三元材料Li[Ni1/3Co1/3Mn1/3]1–xRexO2 (0 ≤ x ≤ 0.04)的稀土掺杂比例则小于4%[19]. 尽管我们模型中的稀土掺杂比例略大于实验的结果, 但在同一量级上. 因此, 我们理论计算的结果与实验具有可比性. 此外, 对于1-2, 1-3, 1-4三条迁移路径, 其对应的能量势垒呈现出明显的方向性, 即由最靠近稀土离子的Li位(1号位)向其他的Li位迁移, 势垒较低, 反之, 从其他Li位向1号Li位迁移, 势垒极大. 这说明Li离子倾向于向远离稀土元素的方向迁移. 进一步地, 在考虑实际迁移过程中, Li离子将会绕过稀土掺杂位而进行迁移.
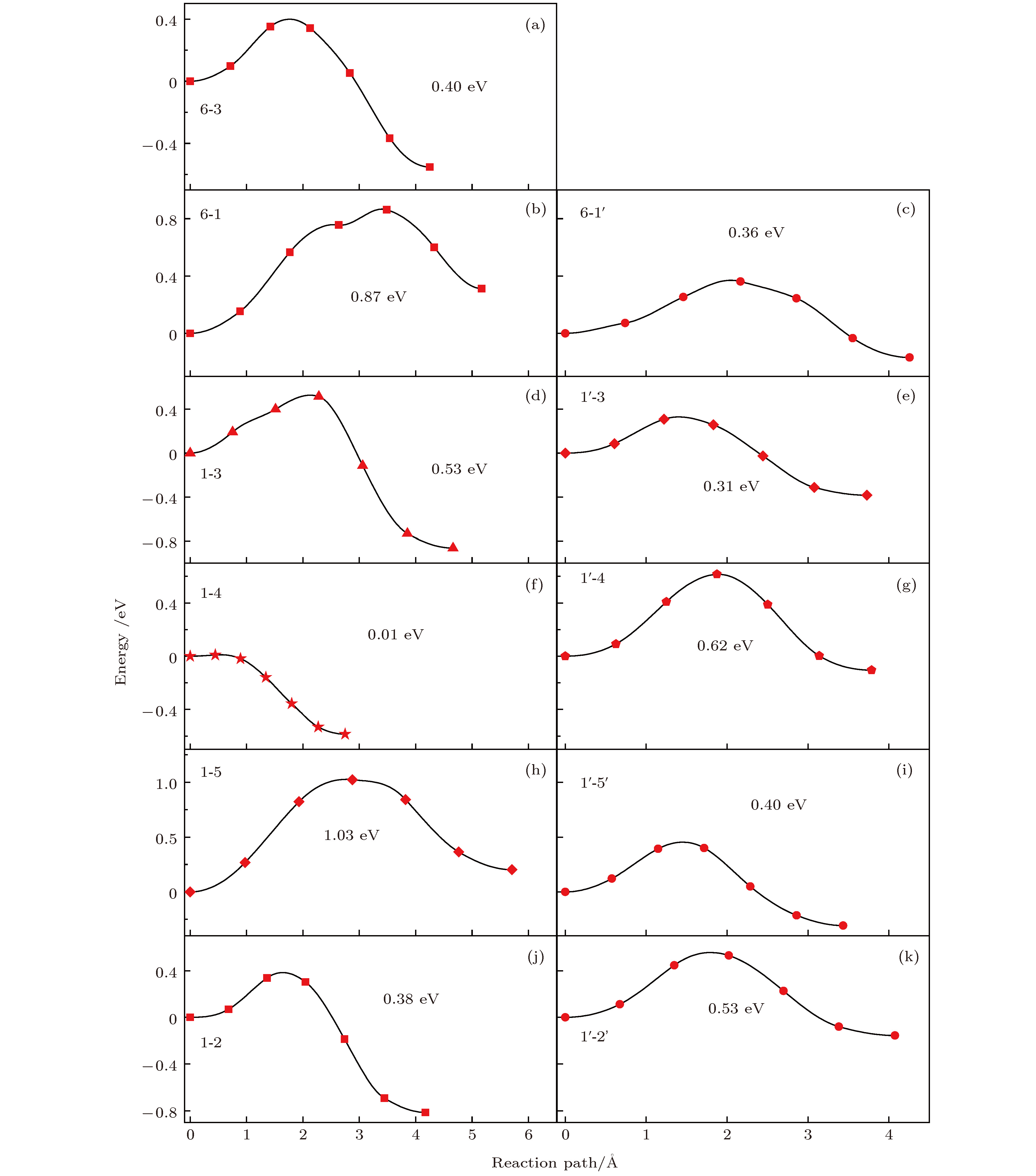 图 4 Li离子在La掺杂Li2MnO3中的迁移势垒, 所有的迁移路径与表3中所列的一致
图 4 Li离子在La掺杂Li2MnO3中的迁移势垒, 所有的迁移路径与表3中所列的一致Figure4. Diffusion energy barriers of Li ions in La-doped Li2MnO3. All diffusion paths are consistent with those listed in Table 3
Li离子在Ce掺杂的Li2MnO3中迁移与La掺杂的情况类似, 迁移势垒如图5所示. 同样的wyckoff位的迁移, 远端位的迁移势垒变化范围为0.31—0.69 eV, 低于未掺杂的情况(0.51—0.84 eV)[37]. 在远离稀土离子处迁移势垒呈现出不同程度的减小, 原因是O和O层间距离较未掺杂时增加, 减小了Li离子迁移时的阻碍. 对于近邻位的离子迁移, 势垒变化范围为0.31—0.89 eV. 其中

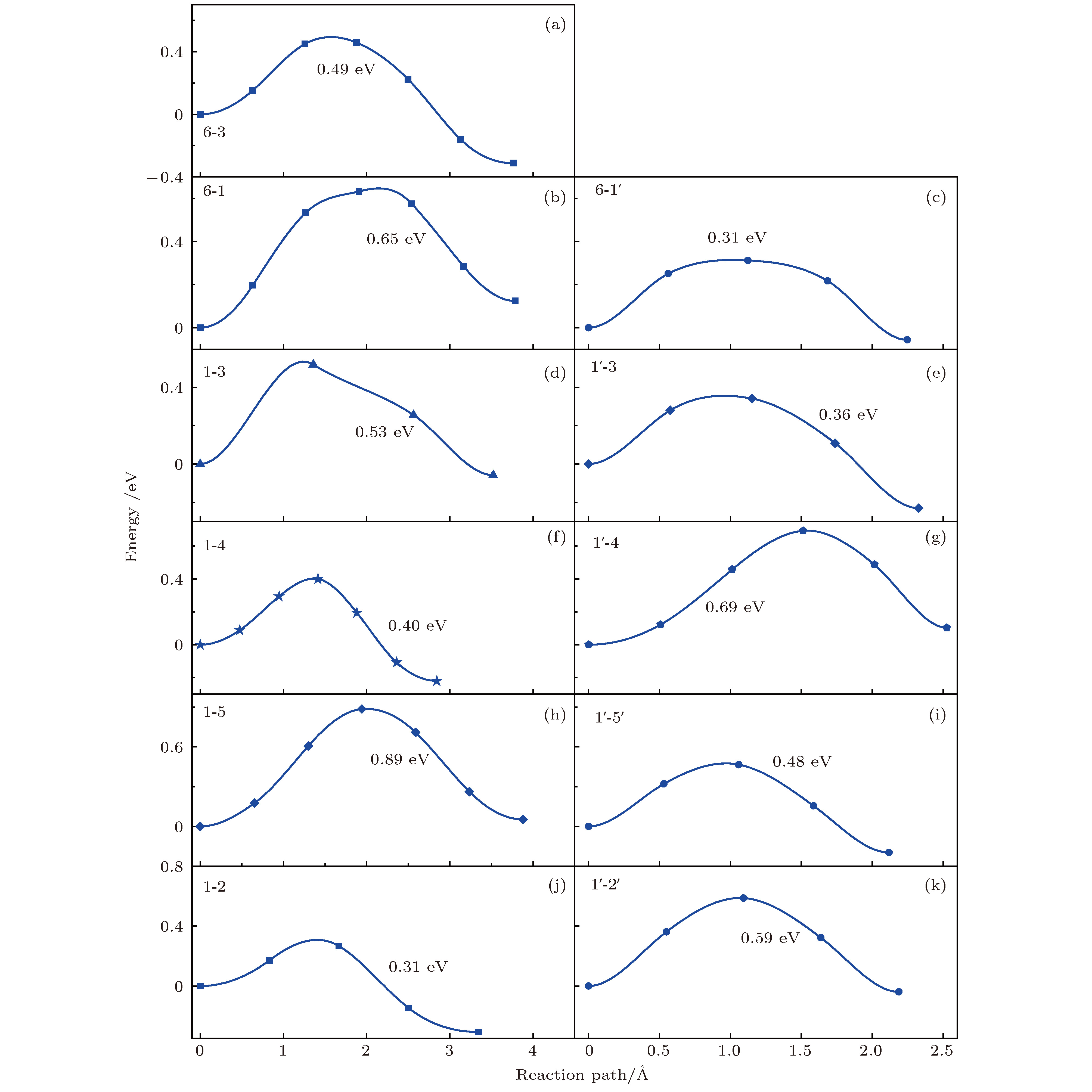 图 5 Li离子在Ce掺杂Li2MnO3中的迁移势垒, 所有的迁移路径与表3中所列的一致
图 5 Li离子在Ce掺杂Li2MnO3中的迁移势垒, 所有的迁移路径与表3中所列的一致Figure5. Diffusion energy barriers of Li ions in Ce-doped Li2MnO3. All diffusion paths are consistent with those listed in Table 3
