 ,*,?,2)
,*,?,2)DETAILED ANALYSIS OF VIBRATIONAL STATES OF OXYGEN IN HIGH TEMPERATURE NON-EQUILIBRIUM FLOWS1)
Hong Qizhen*,?, Wang Xiaoyong*, Sun Quanhua,*,?,2)通讯作者: 2) 孙泉华,研究员,主要研究方向:稀薄气体与非平衡流动. E-mail:qsun@imech.ac.cn
收稿日期:2019-06-4接受日期:2019-09-3网络出版日期:2019-09-03
| 基金资助: |
Received:2019-06-4Accepted:2019-09-3Online:2019-09-03
作者简介 About authors

摘要
高超声速流动在头激波压缩后常处于高 温条件下的热化学非平衡状态. 本文采用态-态方法和双温度模型计算分析了一维正激波后和高超声速钝体绕流驻点线上的氧气热化学非平衡流动. 态-态方法将氧气的每个振动能级当成独立的组分,通过耦合 Euler 方程或驻点线上的降维 Navier-Stokes 方程,数值求解得 到了高温流动中的精细热化学非平衡状态. 而双温度模型假设氧气的振动能级服从 Boltzmann 分布,通过求解振动能方程得到振动温度. 一维正激波后热化学松弛过程的计算结果表明,态-态计算预测的温度分布和氧原子浓度分布较好地吻合了文 献中的实验结果,而经典的双温度模型的预测结果误差较大,且不同双温度模型的计算结果比较发散. 态-态方法详细地给出了所有振动能级的变化过程. 无论是正激波还是脱体激波后的流场,都是高振动能级首先得到激发;但是数密度大的低振动能级先达到热平衡,而高能级 分子要经过很长距离后才能达到热平衡. 在驻点附近,复合反应生成的氧气分子处于高振动能级,导致高振动能级分子数密度显著高于平衡分布. 计算还发现,经典双温度模型的离解反应速率明显偏离态-态计算结果,无法准确体现振动离解耦合效应对离解反应 速率的影响,但是 Park 双温度模型将离解失去的振动能取为 0.3$\sim $0.5 倍分子离解能是比较合理的.
关键词:
Abstract
Hypersonic flow is usually in a thermochemical nonequilibrium state due to high temperature after the bow shock. In this paper, the state-to-state method and two-temperature models are employed to study the thermochemical nonequilibrium processes of oxygen for a post-shock flow and a flow over a blunt body along the stagnation line. The state-to-state method treats each vibrational energy level of molecular oxygen as an independent species, and predicts the number density of each vibrational level by coupling the Euler equations or reduced Navier-Stokes equations along the stagnation line. The two-temperature models assume that all vibrational levels follow the Boltzmann distribution at a vibrational temperature, and a vibrational energy equation is solved to obtain the vibrational temperature. Simulation results show that the distributions of the temperature and species concentration predicted by the state-to-state method are in good agreement with the available experimental results in the literature, while the classical two-temperature models show large errors and the results of different two-temperature models are scattered. The state-to-state method gives detailed information of all vibrational levels along the streamline. After the normal shock or bow shock, the high vibrational levels are first excited but low levels with large number density will reach thermal equilibrium first, whereas high level molecules reach thermal equilibrium only after a long distance. Near the stagnation point, the recombination reaction produces oxygen molecules that are at high vibrational levels, thus the number density of a high vibration level is significantly higher than that of the equilibrium distribution. It is also found that the dissociation rate of classical two-temperature models deviates from the state-to-state result, which cannot accurately account for the coupling effects of vibration dissociation on the dissociation rate. However, it is reasonable for Park’s two-temperature model to take the vibration energy lost by dissociation to be 0.3$\sim$0.5 times of the molecular dissociation energy.
Keywords:
PDF (12853KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
洪启臻, 王小永, 孙泉华. 高温非平衡流动中的氧分子振动态精细分析1). 力学学报[J], 2019, 51(6): 1761-1774 DOI:10.6052/0459-1879-19-145
Hong Qizhen, Wang Xiaoyong, Sun Quanhua.
引 言
高超声速流动中,由于激波强烈的压缩作用和壁面摩擦作用,头激波后的流场温度明显升高. 高温将导致气体分子的转动能、 振动能和电子能的不同程度激发,有时还可能引起离解复合等化学反应. 这些热化学反应发生的难易和快慢程度取决于相应物理过程的碰撞截面或化学反应速率. 然而,碰撞截面和反应速率依赖于温度和其他物理条件,并不容易确定[1-2]. 特别地,气体分子往往处于一定程度的非平衡状态,准确描述流动过程需要刻画流场中气体分子的详细内能状态及精细的热化学过程.工程上一般采用多温度模型来求解高温气体流动的内能状态及其松弛过程,其中最常采用的是 Park 提出的双温度模型[2]. 多温度模型假设气体各能量模式能级上的粒子分布为该能量模式对应温度下的平衡分布(即对应不同温度下的玻尔兹曼分布),通 过求解对应能量模式的能量方程,获得以不同温度表征的能量非平衡特征. 然而高超声速流动中的气体内能分布经常偏离平衡状态,多温度模型采纳的能量模式内平衡分布的基本假设已不再成立, 模型中引入的经验参数只能近似体现非平衡效应[1],并且这些参数的取值还依赖于人为的经验性设定.
目前,考虑内能能级精细分布的态-态计算方法得到了广泛关注[3]. 该方法将气体内能模式的每一能级当作一个独立 的状态,通过与所有组分方程和流动方程耦合求解,得到分子内能能级的详细分布,从而可以更好地揭示高超声速流动中 的热化学非平衡特性与流场演化规律. 态-态方法将内能状态与气体组分一起作为热化学状态处理,内能能级间的跃迁过程与组分间的化学反应都视为基元反 应过程[14],但是热化学状态的数量远远大于气体组分数量,计算资源需求极大增加. 考虑到气体转动能级的松弛时间远短于振动能级的松弛时间,通常认为:相对于分子振动能的非平衡程度,分子的转 动能与平动能处于热平衡. 此外,电子能需要很高的温度才能有显著的激发效果. 因此目前的态-态计算普遍集中在振动能级的计算与分析,计算中只考虑各振动能级与平动能 (V-T) 的热交换、各振动 能级与碰撞分子各振动能级和平动能(V-V-T)的热交换、以及各振动能级的离解反应.
上述热交换和离解反应过程的定量描述都需要各自过程的碰撞截面或者反应速率系数. Landau 和 Teller[6]最早 给出了V-T反应速率的线性半经验表达式,其中的振动松弛时间需要根据实验结果拟合得到[7]. 为从理论上计算振动松弛时间,Schwartz 等[8]针对共线二体碰撞模型提出了基于量子力学一阶微扰 理论的 SSH 振动模型. SSH 模型在低能级可以给出较好的计算结果,但在高温和高能级情况下的跃迁速率与实验结果相差很大. 为了修正 SSH 模型的不足,Kerner 提出了半经典无微扰的强迫谐振子 (FHO) 模型[9],该模型得到的跃迁速率与量 子化学计算结果符合较好. Adamovich等[10]针对非共线碰撞,引入了分子的非简谐效应和位相因子,且拓展了FHO模型;此外还发展了反应速 率计算的解析模型[11],避免了复杂的积分过程.
需要指出的是,SSH 和 FHO 模型都是基于振子模型发展而来,目前更为精确的反应速率计算方法是准经典轨道理论方 法 (quasi-classical trajectory method, QCT)[12-13]. QCT 方法根据分子碰撞的势能面,通过蒙特卡罗方法积分经典哈密顿方程得到轨线状态,进而统计得到某一反应的反应速率. 分子碰撞的势能面一般通过分子的第一性原理计算得到,计算量较大,而 QCT 的计算也需要消耗大量计算资源,因此 基于 QCT 的速率数据并不容易得到. Esposito 等[12-13]运用 QCT 方法建立了氮气和氧气的诸多基元反应的反应速率数据库,但是仍然缺少 V-V-T 反应速率. 此外,量子经典 (quantum-classical) 轨道计算方法加入了量子效应的修正[14-15],准确度略高于 QCT 方法,但计算量更大.
态-态方法能够计算分子内能能级精细分布的变化过程,但是涉及能级间的大量基元反应,计算资源需 求大,特别是 V-V-T 过程不仅反应数量多而且反应速率系数计算繁琐. 流场计算中,由于反应与流动耦合还需要反复计算反应速率,计算量极大,因此态-态方法目前很少在实际问题中应用. 但对于较简单的问题,态-态方法已经可以给出精细的分析. Adamovich 等[10]成功应用 FHO 模型计算了氮气经过正激波后的松弛过程. 徐丹等[16-17]采用简化的基元反应过程并忽略了 V-V-T 过程的多量子步跃迁,研究了氮气的零维振动松弛过程 和超 声速喷管内的准一维热化学非平衡流动,初步揭示了非平衡过程中的微观振动态分布演化规律. 最近,Hao 等[18]采用 FHO 模型求解建立了氧气的 V-V-T 反应速率数据库,计算了正激波后的振动松弛过程,得到 了与最新实验结果 吻合更好的计算结果.
本文针对高超声速流动的高温非平衡特点,采用解析的 FHO 模型和 QCT 拟合数据,计算 O$_{2}$/O 混合物在典型高超声速 流动中的热化学非平衡精细过程. 选择的流动为强激波后的一维非平衡流动和三维钝体绕流问题的驻点线流动,具体分析并比较了态-态计算 方法与经典双温度模型在反应速率上的差异,详细讨论了钝体流场头激波后与近壁面附近的氧分子振动态的非平衡分布 特征,得到了驻点线上非平衡振动-离解耦合的反应速率和等效离解振动能数值.
1 振动非平衡与振动-化学耦合模拟方法
1.1 态-态方法
本文研究 O$_{2}$/O 混合物的高温非平衡流动,其中氧分子的每一振动能级在态-态计算中视作独立的气体组分, 耦合求解所有振动能级的数密度控制方程,从而得到振动能级的精细分布.首先需要确定氧分子的振动能级数目. Silva 等[4]利用量子力学中RKR办法推出处于电子能基态的氧分子共有47个振动能级,$i$ 能级相应的振动能 (单位为焦耳) 为
态-态方法中,处于各振动能级的分子均有机会参与振动跃迁和离解复合反应,这些基元反应包括 振动-平动 (V-T) 传能
振动-振动-平动 (V-V-T) 传能
以及离解-复合反应
这里的离解-复合反应与分子的振动能级有关,即考虑了振动-离解的耦合效应. 非平衡时的氧气 离解的表观反应速率常数可以定义为
其中$k_{f,i}$ 是振动态为$i$的氧分子的离解反应速率常数. 综合热化学过程 (2)$\sim$(4) 以及所有基元反应的正逆反应速率 常数,可以得到各振动能级的氧分子数密度变化表达式
其中,$k_{\rm V - V - T}$ 和 $k_{\rm V - T}$ 分别为 V-V-T 和 V-T 过程的反应速率常数,正逆反应速率满足微观的细致平衡原理. $k_{\rm dis}^{{\rm O}_2 } (i)$ 和 $k_{\rm rec}^{{\rm O}_2 } (i)$ 分别是振动能级 $i$ 的离解和复合速率常数,$[:]$ 代表某一组分 (位于不同振动能级的氧分子已视为独立组分)的分子数密度.
态-态计算得到的是各振动能级的分子数密度,据此可以得到气体的振动能和振动温度. 气体振动能根据气体分 子数密度比例计算
气体振动温度通常指的是根据气体振动能计算得到的平衡振动温度,即
但是平衡振动温度无法体现各振动能级的真实非平衡分布,因此也可以计算振动激发态的振动温度,即根据振动激发态与基态
的数密度比来计算激发态的振动温度
1.2 双温度模型
双温度模型是工程上常用的非平衡近似模型. 通常假设气体的平动能和转动能处于同一温度(即平动温度 $T$) 下的平衡分布, 振动能和电子激发能处于另一温度(即振动温度 $T_{\rm v}$) 下的平衡分布. 化学反应计算时考虑了两个温度的耦合影响,即通过引入非平衡因子 $Z(T,T_{\rm v})$ 来修正化学反应速率常数其中,下标 f 表示离解反应,$k_{\rm f,eq} $ 为气体处于平衡态时的反应速率,$\theta _{\rm d} $ 为离解特征温度. 针对非平衡因子的建模,目前已发展了众多的双温度模型,主要包括:
(1) Park 离解模型[27]:Park 提出离解反应受控于反应控制温度 $T_{\rm a} = T^qT_{\rm v}^{1 - q} $,这里 $q$ 为经验常数,一般取为 0.5 或 0.7,非平衡因子为
(2) Beta 离解模型[30]:假设分子为截断简谐振子,振动态的分布仍然为玻尔兹曼分布,但用有效振动能 $\left( {\theta _{\rm d} - \beta T} \right) R$ ($\beta $ 为模型参数,取为 1.5;$R$ 为单位质量气体常数)来代替单位质量离解能 $E_{\rm d}$,非平衡因子为
(3) Treanor-Marrone 离解模型 (CVDV)[31]:认为处于高振动能级的分子更容易发生离解反应,属于优先离解模型. 同时假设离解反应不影响振动态的玻尔兹曼分布,此时耦合非平衡因子为
其中,$Q$ 为振动配分函数[36],$U$ 为模型参数,本文取为 $\theta _{\rm d} $ 的 $1/3$.
(4) Hammerling 离解模型[32]:等价于 CVDV 模型中模型参数取为 $U = \infty$,表示离解反应发生在分子各个振动能级上的概率是一样的.
(5) Macheret-Fridman 离解模型[34]:认为存在两种基本离解机理,第一是从高振动能级直接离解,这一过程依赖于高 的振动温度;第二是高的相对平动能使得低振动能级分子直接离解,依赖于高平动温度. 耦合非平衡因子为
其中,$T_\alpha = \alpha T_{\rm v} + \left( {1 - \alpha } \right)T$, $\alpha = \left( {\dfrac{m_{\rm A} }{m_{\rm A} + m_{\rm M} }} \right)^2$,并且 $L$ 在不同碰撞对中取不同值. 对于原子- 分子碰撞
分子- 分子碰撞
(6) Kuznetsov 离解模型[28]:认为离解反应仅发生在离解能附近的振动能级,低振动能级通过单量子跃迁到高能级 后才能离解,采用有效振动能级为 $E_{\rm v}^ * = nR\theta _{\rm v}$ ($n$ 为模型参数)来代替离解能 $E_{\rm d} $,耦合非平衡因子为
需要注意的是,振动离解耦合效应除了体现在化学反应速率常数的修正,还包括离解(或复合)反应失去(或获得)振动能的修正. 双温度模型由于不考虑振动能级的非平衡跃迁,通常采用等效的振动能来代替离解失去的振动能. 而态-态计算直接得到每一能级的数密度变化率,通过加权求和可得到准确的振动能变化值. 这一振动能源项可以表示为
双温度模型求解振动能方程得到振动温度,振动能源项除了前面提到的化学反应源项 $\varOmega_{\rm vd} $,还包括振动能 与平动能之间的能量松弛源项[4]
其中,$E_{s,\rm vib} $ 为 $s$ 组分单位质量的振动能,$\tau _s $ 为不同组分的振动松弛时间,本文采用 Millikan 和 White[7] 给出的表达式,并考虑高温修正[1].
1.3 基元反应速率
态-态计算需要得到不同温度下所有态-态过程的基元反应速率. 解析无微扰的半经典 FHO 模型提供了计算双原子分 子振动松弛过程反应速率的办 法[9-10]. 该模型考虑了多振动量子数的跃迁效应,更准确地给出振动松弛反应速率常数. 当粒子相互作用势能为排斥势或 Morse 势能时,可以得到解析的反应截面表达式[9-10],然后通过数值积分给出反 应速率[20]其中,$Z$ 为碰撞频率,$u$ 为碰撞粒子对相对运动速率,$\sigma $ 为反应截面. 但是这一数值积分计算比较耗时,将其应用于复杂的多维流动问题并不可取. 为了提高计算效率,本文 采用 Adamovich 等提出的 V-T 和 V-V-T 反应速率常数的解析表达式[20],用最速下降积分来代替直接数值积分. 对于本文采用的 Morse 势,即 $V\left( r \right) = E\left[ {1 - \exp \left( { - \alpha r} \right)} \right]^2$,该解析模型得到的 V-T 过程反应速率常数为
其中
式中,$i$,$f$ 分布代表初态和终态的振动能级;$\tilde {m},\mu $ 分别为碰撞粒子和振子的约化质量;对于同核双原子分子 $\gamma = 0.5$,$S_{\rm VT}$ 为三维修正因子,$\omega $ 为振子频率,$u_{m0} $ 为相对碰撞速度,$h$ 为普朗克常数,$C_{\rm VT}$ 通过对 式(27)进行迭代求解得到,其中对于原子-分子碰 撞 $z =0$,对于分子-分子碰撞 $z =1$.
类似地,V-V-T 过程反应速率为
式中,$\varOmega $ 为两碰撞分子的振子频率差,$S_{\rm VV}$为三维修正因子,$C_{\rm VV}$ 迭代求解 式 (31) 得到.
解析 FHO 模型表达式 (23) 和式 (28) 形式上较为复杂,但其本质上是代数的解析表达式,计算量不大,因此容易与流动方程耦合求解.
本文计算比较了这一解析 FHO 模型表达式 (present FHO) 与直接 对式 (22) 数值积分的 FHO 模型 (full FHO) 计算 O$_{2}(i)$+O$_{2}(i)$ 的 V-V-T 反 应速 率常数. 图1 给出了振动能级 (1,0) 跃迁到 (0,0) 以及 (2,0 ) 跃迁到 (1,0) 的 FHO 计算结果,同时给出了量子经典轨道计算结果 (QC)[14]. 可以看出,简化的解析 FHO 模型在目前温度范围内与积分 FHO 结果以及 QC 结果一致,表明解析 FHO 模型在提高计算效率的同时, 保证了计算精度.
图1
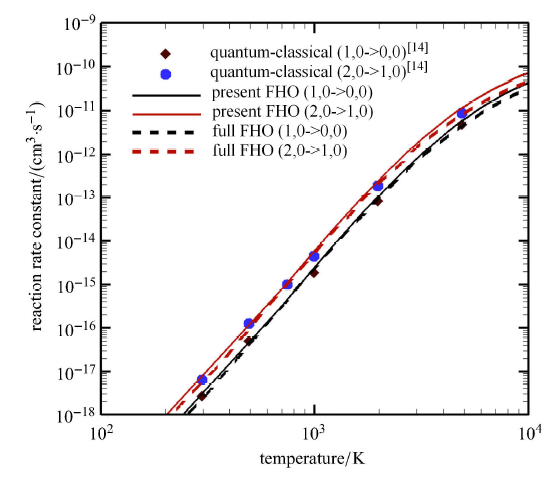 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同模型计算得到的O$_{2}(i)$+O$_{2}(i)$的V-V-T反应速率常数对比
Fig.1Comparison of reaction rate constants of V-V-T reaction O$_{2}(i)$+O$_{2}(i)$ calculated using different models
前面已经提到,准经典轨道 (QCT) 方法能够给出更精确的基元反应速率常数. 文献中已有 O$_{2}(i)$+O 的离解反应、V-T 反 应以及 O$_{2}(i)$+O$_{2}$ 离解反应的反应速率常数QCT计算数据库[13,28-29]. 本文综合 FHO 模型与 QCT 结果,得到了高温 O$_{2}$/O 混合物的所有振动态反应速率常数. 具体 为,O$_{2}$-O$_{2}$ 的离解反应和 V-T 反应的反应速率常数采用拟合的 QCT 数据公式[22],V-V-T 速率常数通过简化的解析 FHO 模型计算;O$_{2}$-O 的离解反应和 V-T 反应的反应速率常数采用 拟合的 QCT 数据公式[11].
2 流动控制方程
2.1 一维正激波后的流动控制方程
气体经过正激波后的热化学松弛过程一般采用一维欧拉方程组来描述,本文考虑的 O$_{2}$/O 气体混合物把位于 47 个振动能级的氧气分子分别视为不同的组元,因此控制方程为其中,$\rho _s $ 为组分密度,$\dot {R}_s $ 为各组分反应源项. 假设激波内没有发生振动松弛与化学反应,激波后气体状态(即松弛初始边界)通 过 Rankine-Hugoniot 关系计算. 为克服计算中可能出现的“刚性”问题,上述方程组采用隐式后向差分法推进求解.
2.2 驻点线模型
由于精细的态-态计算需要大量的计算资源,且求解过程中容易出现“刚性”问题,此前的态-态计 算通常只应用于零维[17]或一维正激波问题[12],难以体现飞行条件下的热化学非平衡特点. 为此,本文将态-态方法与驻点线模型[19] (stagnation line model, SLM) 相结合,计算高超声速钝体绕流问题的驻点线流动.驻点线模型针对高超声速钝体绕流中的高温真实气体效应最显著的驻点线,通过简化三维 Navier-Stokes 方程组,得到在驻点线上 与原方程高度一致的降维模型[31-32]. 结合驻点线模型与不同层次的物理化学模型可以分析不同条件下的钝体绕流的热化学非平衡特性,一方面可以大大降低三维问题 的计算量,另一方面可以把更精细的物理化学模型应用到实际问题的分析研究中.
针对本文研究的 O$_{2}$/O 气体混合物钝体绕流过程,态-态方法耦合驻点线模型在球坐标下的控制方程如下. 动量方程
其中,$v_{1}$ 为径向速度(即沿着驻点线方向),$\rho $ 为密度,$p$ 为压强,$\mu $ 为黏性系数. 总能量方程
其中,$h$ 为焓值,$\rho _m $为 O$_{2}$ 总密度,$D$ 为扩散系数,$\kappa $ 为热传导系数. O$_{2}$ 各振动能级组分方程
以及 O 组分方程
其中,$c$ 为组分质量分数,$\dot {R}$ 为组分反应源项.
分子振动能级组分方程中除包括氧分子与氧原子间的扩散,也包括氧分子与氧分子不同振动能级间的扩散. 扩散过程用 等效二元扩散来描述,等效二元扩散系数采用 Gupta 混合定律[23]计算. 不同振动能级间的等效扩散系数为[24]
其中,$x$ 为组分的摩尔分数,$\vartheta $ 为碰撞积分[24]. 此外,混合物总的黏性系数、热传导系数以及平动、转动热传导系数分别为[23]
式中,$m_s $为粒子质量,$\gamma _s $ 为摩尔浓度,其中碰撞积分项采用 Gupta 拟合曲线公式[23]得到.
上述驻点线模型不考虑周向动量方程从而将方程的维数降为一维,同时引进驻点线上质量流量线性分布的假设[19,37],方 程得以封闭求解. 驻点线模型耦合态-态算法的控制方程组为常微分方程组,可通过有限差分法离散求解. 驻点线的计算区域为头激波至驻点. 假设头激波为强间断,忽略激波内的松弛过程,考虑激波后的黏性梯度,采用激波滑移条件 (shock slip condition) 直接对 N-S 方程在激波前后进行积分[25],得到激波前后气体物理量的关系式. 无量纲化的激波后条件为
其中,上标 $*$ 表示无量纲量,下标 $s$ 表示在激波后取值,$\varepsilon ^2 = \dfrac{1}{Re}$,$Re$ 为来流雷诺数.
3 正激波后的热化学非平衡流动过程分析
本文首先应用态-态方法计算氧气过正激波后的热化学松弛过程,一方面分析正激波后的非平衡流动特性,另一 方面检验态-态计算的可靠性. Ibraguimova 等[19]通过吸收光谱中 Schumann-Runge 光谱带的分析得到了正激波后振动温度和组分浓度分布. 本节选取其中总焓最 大的一组实验条件进行计算,控制方程为方程组 (32),来流条件为 $V_{\infty } =4440$m/s,$P_{\infty } =106.658$Pa,$T_{\infty } =295 $K.图2给出了态-态计算得到的激波后各物理量随松弛距离的变化曲线. 值得注意的是,来流氧气经激波压缩,气体平动温度远大于振 动温度(振动温度经激波不变). 沿激波波后方向,氧分子通过V-T传能使得振动温度上升、平动温度下降;同时高温还使得氧分子发生离解,氧原子质量分数增大, 平动温度进一步下降. 由于波后压力变化相对较小,根据状态方程,平动温度下降使得气体密度上升;由于质量守恒,密度上升使得气体速度下降;根据 动量守恒,速度下降使得压力有所增加. 上述变化,随着松弛距离的增大,变化趋势逐渐放缓,最终达到平衡状态.
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2态-态方法计算得到的各物理量随正激波后距离的变化曲线
Fig.2Calculated curves of physical quantities behind a normal shock wave using the state-to-state approach
图2还给出了由式 (8) 定义的能量表征的平衡振动温度(图中 $T_{\rm v,eq}$) 与由 式 (9) 定义的第一振动激发态的振动温度 (图中 $T_{\rm v,1}$). 两种振动温度的变化趋势基本一致. 但是在激波附近,由于处于第一振动激发态的分子不仅得到基态分子跃迁的补充,而且向高能级激发而损失,使得计算得到的第 一振动激发态的振动温度低于能量表征的平衡振动温度. 此后由于高振动能级的分子更容易发生离解反应,平衡振动温度低于第一振动激发态的振动温度. 而随着松弛距离的进一步增大,振动温度趋于平动温度,不同定义的振动温度也就不存在差别了.
为了详细分析松弛过程中的振动能级分布变化,图3给出了5个位置的归一化振动能级的数密度分布. 在激波处 ($x=0$),由于 热化学反应尚未开始,振动能级处于低温状态下的平衡分布. 通过激波后,由于振动能远小于平动能,V-T 和 V-V-T 过程首先发生. 激波后 ($x=0.09$, 0.29mm) 的流场特征是,振动松弛明显,离解反应处于诱导期,因此高能级的分子数密度显著大于玻尔兹曼分布的数密度(过分布). 此后,随着高能级的分子数密度逐步积累,离解反应开始发生并占据主导地位,这一区域内 ($x=0.99$, 9.99mm) 的高能级分子数密度明显低于玻尔兹曼分布(欠分布). 这是由于离解反应消耗了大量高能级分子,而此时平动能与振动能趋近相等,无法补充足够的高能级分子.
图3
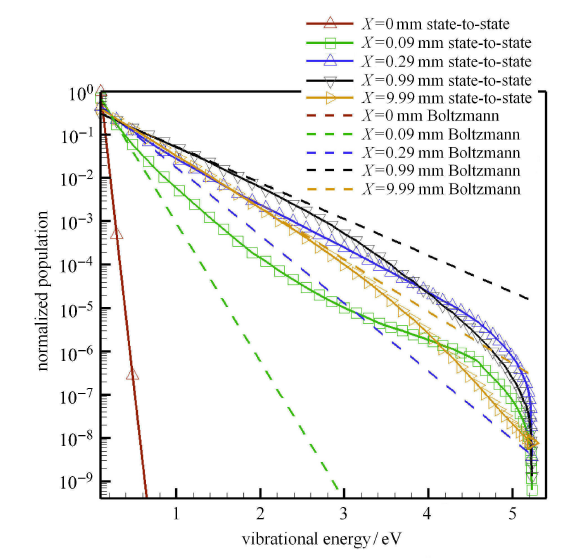 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3激波后不同位置振动能级归一化数密度分布
Fig.3Distribution of normalized number density of vibrational energy levels at different positions
图4给出了不同振动能级归一化数密度随着波后距离的变化. 可以看出,图中显示的所有能级 (v2-v46) 均首先经历 V-T 传能过 程导致归一化数密度逐渐增大,并显著偏离玻尔兹曼分布,以较高能级尤为显著. 另外,低振动能级与平动能的松弛快,更短距离内趋近于玻尔兹曼分布,而高能级在随后的离解反应过程中被大量消耗. 因此,气体中数密度大的低能级首先达到玻尔兹曼分布,呈现出热平衡状态;而高能级分子与化学反应耦合明显,要经过很 长的松弛距离后才能达到热平衡状态,此时气体趋近于真正的热化学平衡态.
本文还采用常用的 6 种双温度模型预测热化学非平衡松弛过程. 图5 和图6 分别给出了预测的正激波后温度和氧原子质量分数结果,其中含态-态计算结果、 4种基于双温度假 设的耦合模型计算结果以及文献[13] 中的相关实验结果. 可以看出,态-态计算结果与实验数据符合较好,尤其是氧原子质量分数分布更接近实验数据. 双温度模型结果依赖于模型的选择,预测结果比较发散.
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同振动能级归一化数密度在激波后的分布
Fig.4Distribution of normalized number density of different vibrational energy levels behind shock wave
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5不同模型预测的正激波后温度分布比较
Fig.5Comparison of temperature distributions behind shock wave calculated using different models
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6不同模型预测的正激波后氧原子质量分数分布比较
Fig.6Comparison of mass fraction distributions behind shock wave calculated using different models
从图5 和图6 可知,在靠近激波后区域,各双温度模型计算得到的V-T松弛速率都比态-态计算结果大,振动温度上升很快,导致振动-离解耦合反应速率偏大 (Macheret-Fridman 模型[34] 除外),波后氧原子质量分数显著大于实验结果. 其中,Park 模型[27]预测的振动温度存在明显的过冲现象(振动温度大于平动温度),这是由于模型中的振动离解耦合能量源项(即式 (20)) 采用了不合理的假设导致的,即简单地将离解失去的振动能设为平均振动能. Kuznetsov 模型[28] 与 Beta 模型[30](图中没有显示)得到类似的结果,但过冲相对较小,它们均采用有效振动能级来代替离解失 去的振动能,属于简化的优先离解模型. 值得注意的是,图5 给出的态-态结果是基于第一振动激发态的平衡振动 温度,局部区域存在该温度大于平动温度的情况,但从图5 可知,态-态预测结果没有过冲现象. CVDV 模型[31] 与 Hammerling 模型[32] (图中没有显示,Hammerling 模型反应速率稍大)假设高能级振子的离解速率远大于低能级振子,预测得 到的离解反应速率最大,剧烈的离解反应导致平动温度、振动温度整体偏低. 此外,Macheret-Fridman 模型[34]离解速率较低,能较 好地捕捉激波后的离解反应诱导区,但达到整体的热化学平衡所需的松弛距离最长,远离激波处的氧原子质量分数偏低. 总的来说, 上述多种双温度模型都未能很好地再现激波后的非平衡过程,且模型中都需要引入经验参数,因此存在着很大的不确定性.
图6 还比较了 Hao 等[18]采用态-态方法得到的氧原子质量分数计算结果. 文献[12] 是对式 (22) 直接进行数值积分得到 FHO 模型的反应速率常数,计算量较大,但与本文使用的解析 FHO 模型 (即 式 (23)、 式 (28)) 结果差别不大. 总体来讲,本文态-态计算中运用的基元反应速率常数能够详细描述热化学非平衡过程,并能较好地复现实验结果.
4 高超声速钝体绕流驻点线的非平衡分析
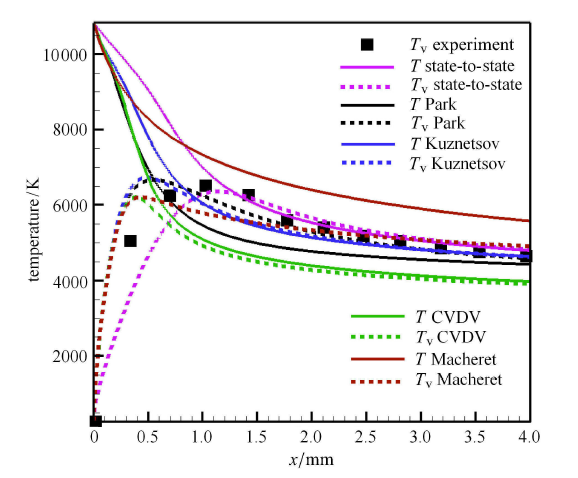
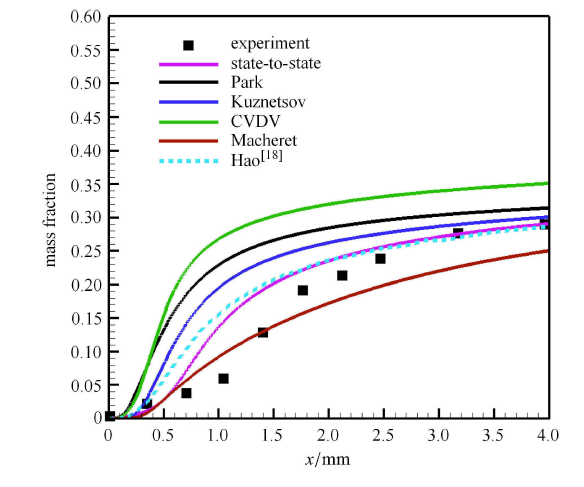
高超声速钝体绕流是高温热化学非平衡现象比较突出的典型问题,开展三维全流场复杂外型的态-态计 算需要很大的计算量,难以 在现阶段内实现. 但态-态方法与驻点线模型相结合,可以得到高温激波层内关键的驻点线流动参数, 能够分析驻点线上的精细热化学非平衡过程, 计算量也是工程上可以接受的.考虑球头半径为 0.1m 的钝体流动,其中氧气来流参数 $V_{\infty } =4440$m/s,$P_{\infty }=106.658$Pa,$T_{\infty }=295$K,与上节中正激波前来流参数一致,球头壁面温度 为$T_{w}=300$K. 图7 给出了态-态计算得到的驻点线上各物理量变化曲线,同时比较了广泛使用的 Park 模型[27]和 CVDV 模型[31] 的模拟结果. 由于不同模拟预测的激波脱体距离有一定差异,为了更直观地体现物理化学模型对振动温度和组分质量分数的影响,图中 横坐标用激波脱体距离进行了归一化处理. 计算发现,不同模型得到的压力、速度值差别不大,因此图7(a) 只显示了态-态计算结果. 但不同模型对平动温度与密度的预测有较大影响,主要是因为模型决定了化学反应速率和反应热的大小. 图7(b) 给出了不同模型预测的振动温度和氧原子质量分数. 在激波后,Park 模型[27]和 CVDV 模型[31]的离解反应速率远大于态-态计算得到的反应速率, 这与前文正激波后的流动规律保持一致. 在壁面附近, 态-态计算的复合反应速率也比两种双温度模型结果小,所 以氧原子质量分数以及振动温度预测值均大于双温度模型结果.
图7
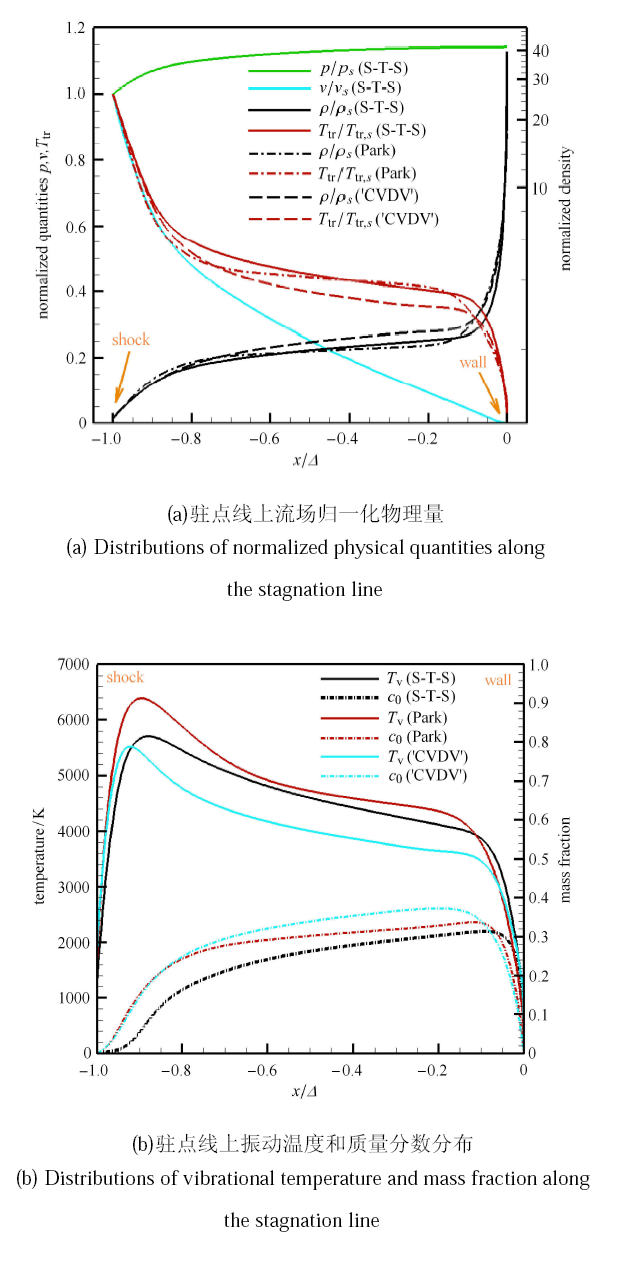 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7不同模型驻点线上各物理量变化曲线
Fig.7The curves of physical quantities along the stagnation line
为了分析驻点线上的能量非平衡特征,图8 给出了驻点线上的温度分布,包括平动温度和第 1,5,10,15,20,30,40,45 振动激发态的振动温度分布. 可以看到,在激波后小区域内,由于高能级分子数密度通过 V-T 传能相比于未激发时急剧增大,振动温度上升很快;而低能级振动温度上升相对较慢. 但是马上随着解离反应的进行,高振动能级的振动温度不再增大,甚至还略微减小,与平动温度相差明显;而低振动能级温度主要受V-T传能影响,逐渐与平动温度趋向一致. 在近壁面附近,由于温度边界层的存在,边界层内的低温引起强烈的复合反应,复合反应生成的分子具有较高的振动能级, 使得高振动能级分子数密度明显增大,超过了高能级退激的数密度,因而高能级振动温度在壁面附近上升;而低振动能级分子受复合反应影响小,与平动温度保持平衡. 图中还显示了能量表征的平衡振动温度,该振动温度与基于第一振动激发态的振动温度相近,说明低能级的粒子数占据了绝大部分.
图8
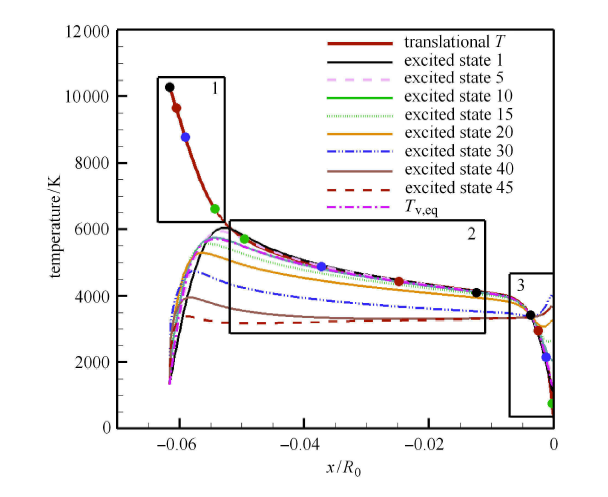 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8驻点线上平动温度、各能级振动温度和平衡振动温度分布
Fig.8Temperature distributions of different vibrational energy levels along the stagnation line
态-态计算给出了驻点线上所有振动能级的数密度,因而可以分析驻点线上各振动能级的演化规律. 为此,在驻点线上靠近 激波后、中间区域以及近壁面处各取4个观察点(如图8 中用黑框标志的 1, 2, 3 区域,其中不同颜色标志的位置点与图9 中结果一一保持对应), 得到不同位置的振动能级数密度分布,并与第一激发态的振动温度下的玻尔兹曼分布进行对比.
图9
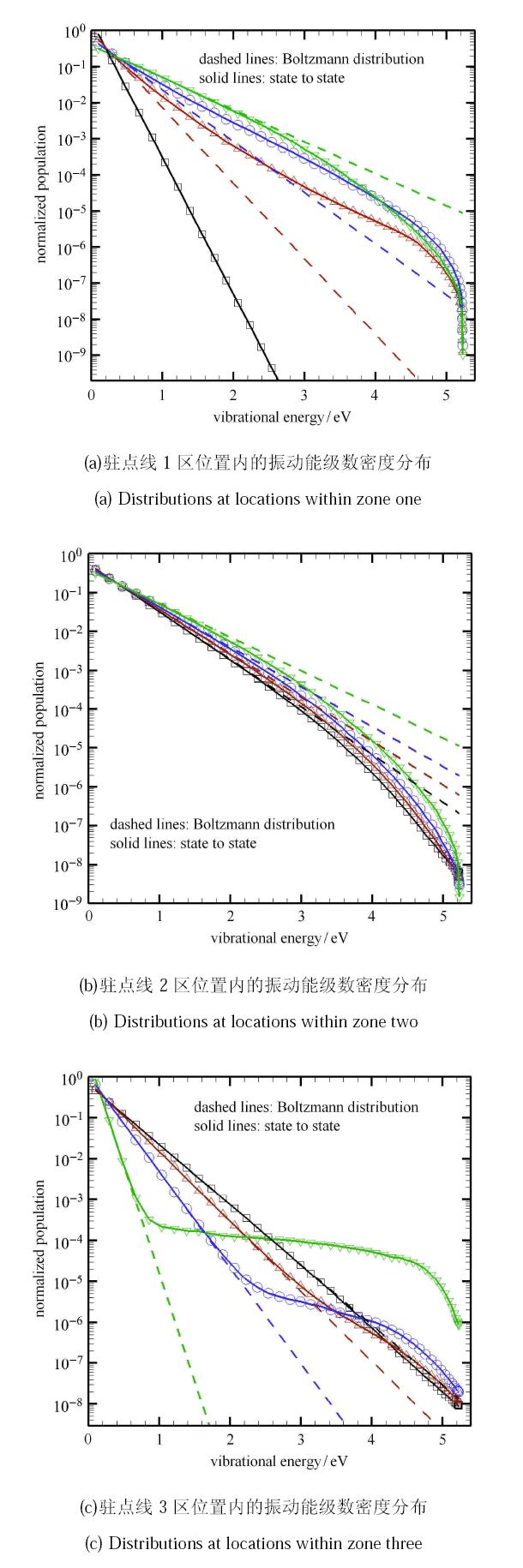 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9驻点线关键位置的振动能级数密度分布
Fig.9Number density distribution of vibrational Energy levels along the stagnation line
图9(a) 给出驻点线上激波后较近距离处的振动能级数密度分布. 图中黑线为激波后初始位置的平衡分布. 激波后显著的平动-振动传能 过程使得振动能级极大地偏离玻尔兹曼分布. 振动能激发使得高能级的数密度明显大于玻尔兹曼平衡分布,呈现“过分布”状态(如图 9(a) 中的红线和蓝线). 当高能振子数密度积累到一定程度后,离解反应开始发生并逐步增强,反应诱导区后的高能级振子优先离解,振动能被大量消耗, 此时高能级分子数将明显低于玻尔兹曼分布,呈现“欠分布”状态(如图9(a) 中的绿线).
图9(b) 为驻点线中间区域处的振动能级数密度分布,此处分子离解反应占主导,高能级振子被大量消耗,4个观察点均 呈现“欠分布”状态. 但越靠近壁面,“欠分布”状态越弱.
图9(c) 为驻点线近壁面区域处的振动能级数密度分布. 由于壁面为低温条件,越靠近壁面的气体温度越低,原子复合效应越 显著. 最低几个能级的振子温度与平动温度平衡较快,基本按照平动温度大小呈玻尔兹曼分布;而高振动能级从原子复合反应得到的数 密度大于因低温退激发的数密度,因此高能级极大地偏离玻尔兹曼分布;还有在中间能级区域出现了明显的平台区. 此外,从中间区域的解离反应过渡到近壁面区的复合反应,在近壁面区位置(如图9(c) 中的黑线)还观察到接近平衡的能级分布, 说明来流经激波压缩后的热化学反应到该处达到了平衡.
总的看来,在驻点线上的关键位置,气体处于热化学的强非平衡状态,经典双温度模型采用的振动能级处于玻尔兹曼分布 的假设已不再成立.
前文已经提到,双温度模型处理非平衡问题有两个关键,一是如何在平衡条件下得到的反应速率常数中增加振 动-离解耦合效应(即 式 (图10)),二是如何定义离解反应失去的振动能,也就是振动离解耦合能量源项(即式 (20)).
图10
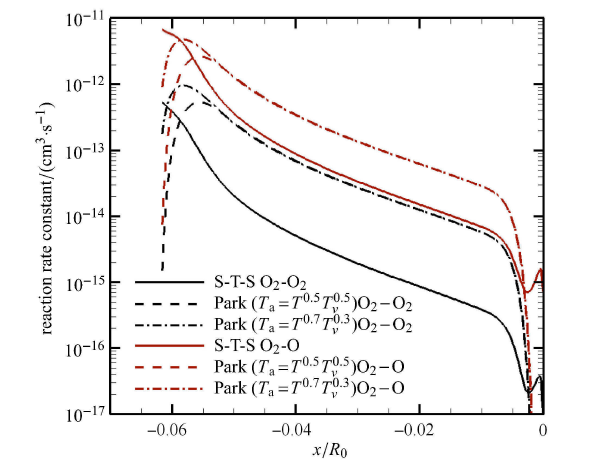 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10驻点线上氧气离解速率常数比较
Fig.10Comparison of oxygen dissociation rate constants along the stagnation line
图10 比较了驻点线上的氧气离解速率,其中实线为态-态计算得到的表观离解速率,虚线为 Park 模型给出的双温度下的离 解速率. 从图可知,态-态计算的离解速率从脱体激波后沿着驻点线逐渐减小;但在驻点附近的温度边界层内,先急剧下降,然 后逐渐增加再减少. 而 Park 模型得到的离解速率,不仅在量级上与态-态计算结果有差异,在驻点线上的整体变化趋势 也不一样,特别在脱体激波后和边界层内呈现两端小的分布,说明Park模型定义的双温度控制参数并不能很好地反应出流动非 平衡对反应速率影响的本质特点.
对于双温度模型中振动能源项处理的评估,以工程中广泛应用的 Park 模型[27] 和 CVDV 模型[31]为例说明. Park 模型[27]假设离解反应失去的振动能为分子的平均振动能或者分子离解能的一部分. 而 CVDV 模型[31] 本质上属于优先离解模型,其振动能源项中离解失去的振动能 $e_{\rm eff} $为
图11给出了不同离解振动能源项随驻点线变化的计算结果. 可以看出,假设离解失去的振动能为分子平均振动能过低地估计了离解振动能源项,因为离解反应倾向发生于高的振动能级. CVDV模型的离解振动能源项大于平均振动能,但也比态-态计算结果低. 态-态计算结果大致处于 0.3$\sim $0.5 倍分子离解能范围,具体数值随驻点线位置有明显变化. 因此假设离解反应失去的振动能为 0.3$\sim $0.5 倍分子离解能,在工程应用上具有参考意义.
图11
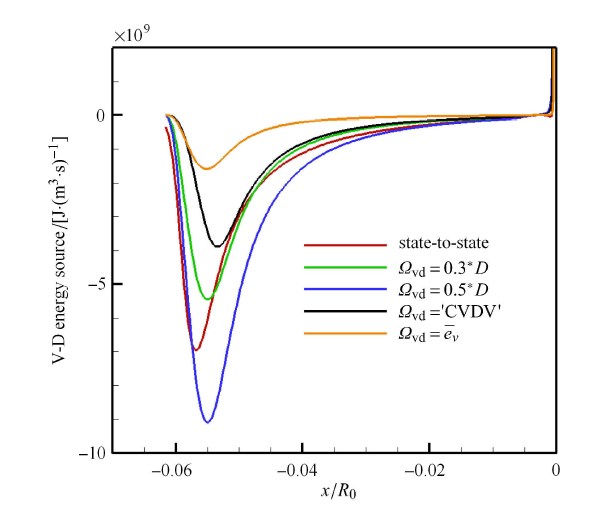 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11不同模型离解振动能源项比较
Fig.11Comparison of lost vibrational energy in different models
以上分析针对的是一种流动状态下的非平衡过程,针对不同流动状态的初步研究发现,驻点线上的关键区域内的振动能级处于 非平衡状态. 譬如,脱体激波后和温度边界层内,分子振动能级显著偏离玻尔兹曼分布. 但在脱体激波距离较大的激波层内也会有局部平衡区域(如图9(c) 中的黑线). 如果激波脱体距离变小(球头半径 0.01m),激波层内流动几乎都处于热化学非平衡状态,驻点线上流场演化规律定性一致,但 非平衡效应更为显著.
5 结 论
本文采用态-态方法和典型双温度模型计算分析了一维正激波后和高超声速钝体绕流驻点线上的氧气高温热化学非 平衡流动. 得到主要结论如下:通过一维正激波后的非平衡流动计算发现,在靠近激波后区域,典型双温度模型计算得到的V-T松弛速率都比态-态计算结果大,
振动温度上升快,波后氧原子质量分数显著大于实验结果. 态-态计算结果相较于双温度模型准确性更高,其温度分布和氧原子浓度分布与实验结果吻合更好.
高超声速钝体绕流在驻点线上的热化学非平衡过程复杂. 在脱体激波后,平动-振动传能过程使得振动能级极大地 偏离玻尔兹曼分布. 高振动能级的数密度明显大于玻尔兹曼平衡分布,呈现“过分布”状态. 当离解反应发生并逐步增强,高能级振子因优先离解被大量消耗,振动能呈现“欠分布”状态. 当化学反应逐渐达到平衡后,振动温度也与玻尔兹曼分布一致. 在温度边界层内,低温下的原子复合反应显著,高振动能级数明显高于玻尔兹曼分布,中间能级区域可能出现平台区. 这种精细的振动能级变化是经典的双温度模型无法预测的. 双温度模型计算的振动-离解耦合反应速率也明显不同于态-态得到的表观反应速率,不能准确体 现振动离解耦合效应对反应速率的影响,因此不同模型对流场温度与密度等的预测有明显影响. 特别地,在激波附近,Park 模型和 CVDV 模型预测的振动温度和氧原子质量分数偏大,而在驻点附近又偏小.
从驻点线流动看,Park 模型得到的离解速率,在脱体激波后和边界层内呈现两端小的分布,与 态-态结果有定性 差异;而在居于中间的大部分区域,Park 结果定性上与态-态结果一致,但数值上近似大一个量级. 对于离解振动能源项,态-态计算表明离解能大致处于 0.3 及 0.5 倍分子离解能的范围内,具体数值随驻点线位置变 化差异明显. Park 双温度模型中将离解反应失去的振动能取为 0.3$\sim $0.5 倍分子离解能是工程应用上较为合理的假设.
态-态计算可以精细地描述各振动态的分布与松弛过程,对离解与复合化学反应是自然耦合的,不需要特别地处理. 常用的双温度模型无法精细描述振动态间的演化规律,且模型中需要引入经验参数,存在着很大的不确定性;此外双温度模型结果 依赖于模型的选择,预测结果比较发散. 因此,精细的态-态计算方法对高温非平衡流动中物理过程的分析和定量描述具有重要意义.
致谢
本文作者感谢俄亥俄州立大学 Igor V. Adamovich 教授以及香港理工大学郝佳傲博士的有益讨论.参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOIURLPMID [本文引用: 3]

A hypersonic vehicle traveling at a high speed disrupts the distribution of internal states in the ambient flow and introduces a nonequilibrium distribution in the post-shock conditions. We investigate the vibrational relaxation in diatom-atom collisions in the range of temperatures between 1000 and 10?000 K by comparing results of extensive fully quantum-mechanical and quasi-classical simulations with available experimental data. The present paper simulates the interaction of molecular oxygen with argon as the first step in developing the aerothermodynamics models based on first principles. We devise a routine to standardize such calculations also for other scattering systems. Our results demonstrate very good agreement of vibrational relaxation time, derived from quantum-mechanical calculations with the experimental measurements conducted in shock tube facilities. At the same time, the quasi-classical simulations fail to accurately predict rates of vibrationally inelastic transitions at temperatures lower than 3000?K. This observation and the computational cost of adopted methods suggest that the next generation of high fidelity thermochemical models should be a combination of quantum and quasi-classical approaches.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
DOIURLPMID [本文引用: 2]
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
URL
化学反应动力学是化学领域最基础的学科之一,量子态分辨的基元化学反应动力学在最为基本的原子与分子的层次上对化学反应的机制提供深刻的理解。该领域的科学家们通过精心设计的实验和高精度的理论计算,使得态态反应动力学在过去的半个多世纪中取得了长足的进步,实验和理论的相互结合极大地促进了我们对化学反应本质的认识。本文从实验研究的角度,通过对实验技术的发展和对H2O光解离、H+H2、F+H2、Cl+H2、OH+H2、F+CH4等具体实例的态态动力学研究的简介,概况介绍了过去二十年里态态化学反应动力学研究所取得的进展,希望借此为读者提供对化学反应动力学领域的一个概略认识。
URL
化学反应动力学是化学领域最基础的学科之一,量子态分辨的基元化学反应动力学在最为基本的原子与分子的层次上对化学反应的机制提供深刻的理解。该领域的科学家们通过精心设计的实验和高精度的理论计算,使得态态反应动力学在过去的半个多世纪中取得了长足的进步,实验和理论的相互结合极大地促进了我们对化学反应本质的认识。本文从实验研究的角度,通过对实验技术的发展和对H2O光解离、H+H2、F+H2、Cl+H2、OH+H2、F+CH4等具体实例的态态动力学研究的简介,概况介绍了过去二十年里态态化学反应动力学研究所取得的进展,希望借此为读者提供对化学反应动力学领域的一个概略认识。
DOIURLPMID [本文引用: 1]
Ultrasound is now a clinically-accepted modality in the management of osteoporosis. The most common commercial clinical devices assess fracture risk from measurements of attenuation and sound speed in cancellous bone. This review discusses fundamental mechanisms underlying the interaction between ultrasound and cancellous bone. Because of its two-phase structure (mineralized trabecular network embedded in soft tissue-marrow), its anisotropy, and its inhomogeneity, cancellous bone is more difficult to characterize than most soft tissues. Experimental data for the dependences of attenuation, sound speed, dispersion, and scattering on ultrasound frequency, bone mineral density, composition, microstructure, and mechanical properties are presented. The relative roles of absorption, scattering, and phase cancellation in determining attenuation measurements in vitro and in vivo are delineated. Common speed of sound metrics, which entail measurements of transit times of pulse leading edges (to avoid multipath interference), are greatly influenced by attenuation, dispersion, and system properties including center frequency and bandwidth. However, a theoretical model has been shown to be effective for correction for these confounding factors in vitro and in vivo. Theoretical and phantom models are presented to elucidate why cancellous bone exhibits negative dispersion, unlike soft tissue, which exhibits positive dispersion. Signal processing methods are presented for separating "fast" and "slow" waves (predicted by poro-elasticity theory and supported in cancellous bone) even when the two waves overlap in time and frequency domains. Models to explain dependences of scattering on frequency and mean trabecular thickness are presented and compared with measurements. Anisotropy, the effect of the fluid filler medium (marrow in vivo or water in vitro), phantoms, computational modeling of ultrasound propagation, acoustic microscopy, and nonlinear properties in cancellous bone are also discussed.
DOIURLPMID [本文引用: 2]
The picosecond optical-optical double resonance experiment in a supersonic free jet as well as the vapor-phase phosphorescence indicates that the decay of T(1) Cl(2)CS belongs to the intermediate case of the classification scheme for electronic relaxation. The A(fast)/A(slow) pre-exponential ratio in the biexponential T(1) decay is much greater under picosecond excitation than under nanosecond excitation. In vapor phase at low pressure, the phosphorescence exhibits a decay time that varies with the coherence width of the laser used for excitation. Both the T(1) and the S(1) decay times of Cl(2)CS depend strongly on temperature, indicating that Coriolis coupling plays an important role in mode mixing (intramolecular vibrational redistribution).
DOIURLPMID [本文引用: 1]
This work presents a theoretical treatment of the vibrational line shape generated in a femtosecond stimulated Raman spectroscopy (FSRS) experiment under conditions in which the probed vibration undergoes a significant frequency shift during its free induction decay. This theory is applied to simulate the FSRS lineshapes previously observed in rhodopsin (Kukura et al. Science 2005, 310, 1006). The previously determined relaxation times for formation of the trans-photoproduct of rhodopsin were calculated using an incorrect equation for the time dependence of the observed frequency shifts. Here the data are reanalyzed by calculation of the corrected frequency sweep occurring during the vibrational free induction decay. It is shown that the calculated frequency shifts and general conclusions of the original work are sound but that the coherent vibrational frequency shifts of the C(10), C(11), and C(12) hydrogen-out-of-plane vibrations occur with a 140 fs time constant rather than the previously reported 325 fs time constant. This time constant provides an important constraint for models of the dynamics of the cis to trans isomerization process.
DOIURL [本文引用: 3]
DOIURL [本文引用: 4]
DOIURL [本文引用: 2]
DOIURL [本文引用: 4]
DOIURLPMID [本文引用: 4]
In this work, a computational model of state-to-state energy flow in gas ensembles is used to investigate collisional relaxation of excited OH, present as a minor species in various bath gases. Rovibrational quantum state populations are computed for each component species in ensembles consisting of 8000 molecules undergoing cycles of binary collisions. Results are presented as quantum state populations and as (approximate) modal temperatures for each species after each collision cycle. Equilibration of OH is slow with Ar as the partner but much faster when N(2) and/or O(2) forms the bath gas. This accelerated thermalization is shown to be the result of near-resonant vibration-vibration transfer, with vibrational de-excitation in OH matched in energy by excitation in bath molecules. Successive near-resonant events result in an energy cascade. Such processes are highly dependent on molecule pair and on initial OH vibrational state. OH rotational temperatures initially increase, but at equilibration, they are lower than those of other modes. Possible reasons for this observation in molecules such as OH are suggested. There are indications of an order of precedent in the equilibration process, with vibrations taking priority over rotations, and potential explanations for this phenomenon are discussed.
DOIURLPMID [本文引用: 3]
The oxygen absorbance was studied at wavelengths 200-270 nm in Schumann-Runge system behind the front of a strong shock wave. Using these data, the vibrational temperature Tv behind the front of shock waves was measured at temperatures 4000-10,800 K in undiluted oxygen. Determination of Tv was based on the measurements of time histories of absorbance for two wavelengths behind the shock front and on the results of detail calculations of oxygen absorption spectrum. Solving the system of standard quasi-one-dimensional gas dynamics equations and using the measured vibrational temperature, the time evolution of oxygen concentration and other gas parameters in each experiment were calculated. Based on these data, the oxygen dissociation rate constants were obtained for thermal equilibrium and thermal non-equilibrium conditions. Furthermore, the oxygen vibrational relaxation time was also determined at high temperatures. Using the experimental data, various theoretical and empirical models of high-temperature dissociation were tested, including the empirical model proposed in the present work.
DOIURLPMID [本文引用: 1]
Vibrational relaxation of O2(X 3sigma(g)-, upsilon=2,3) by O2 molecules is studied via a two-laser approach. Laser radiation at 266 nm photodissociates ozone in a mixture of molecular oxygen and ozone. The photolysis step produces vibrationally excited O2(a 1delta(g)) that is rapidly converted to O2(X 3sigma(g)-, upsilon=2,3) in a near-resonant adiabatic electronic energy-transfer process involving collisions with ground-state O2. The output of a tunable 193-nm ArF laser monitors the temporal evolution of the O2(X 3sigma(g)-, upsilon=2,3) population via laser-induced fluorescence detected near 360 nm. The rate coefficients for the vibrational relaxation of O2(X 3sigma(g)-, upsilon=2,3) in collision with O2 are 2.0(-0.4)(+0.6) x 10(-13) cm3 s(-1) and (2.6+/-0.4) x 10(-13) cm3 s(-1), respectively. These rate coefficients agree well with other experimental work but are significantly larger than those produced by various semiclassical theoretical calculations.
DOIURL [本文引用: 1]
热化学非平衡流模拟中广泛应用的双温度或多温度模型不能描述分子在各振动能级上的分布,只能假设其满足振动温度下的Boltzmann分布。通过采用态-态模型研究非平衡过程中粒子的能级分布特点,有望为改进双温度或多温度模型提供思路。对静止的N2/N气体混合物,在各类不同初始条件和控制温度、压力下,采用态-态模型研究气体的化学组成和分子振动能级分布演化规律,分析各类微观过程的特征与贡献,结果表明:平动-振动能量交换过程起支配作用,促使振动能级分布趋于平动温度下的Boltzmann分布,而振动-振动能量交换过程主要影响能级分布变化的过渡过程特点;离解区和复合区能级分布的变化特点不同;关于非平衡过程中粒子微观分布的研究结果可为改进高超声速非平衡流模拟中的热化学模型提供参考依据。
DOIURL [本文引用: 1]
热化学非平衡流模拟中广泛应用的双温度或多温度模型不能描述分子在各振动能级上的分布,只能假设其满足振动温度下的Boltzmann分布。通过采用态-态模型研究非平衡过程中粒子的能级分布特点,有望为改进双温度或多温度模型提供思路。对静止的N2/N气体混合物,在各类不同初始条件和控制温度、压力下,采用态-态模型研究气体的化学组成和分子振动能级分布演化规律,分析各类微观过程的特征与贡献,结果表明:平动-振动能量交换过程起支配作用,促使振动能级分布趋于平动温度下的Boltzmann分布,而振动-振动能量交换过程主要影响能级分布变化的过渡过程特点;离解区和复合区能级分布的变化特点不同;关于非平衡过程中粒子微观分布的研究结果可为改进高超声速非平衡流模拟中的热化学模型提供参考依据。
[本文引用: 2]
[本文引用: 2]
DOIURL [本文引用: 2]
DOIURLPMID [本文引用: 3]
The oxygen absorbance was studied at wavelengths 200-270 nm in Schumann-Runge system behind the front of a strong shock wave. Using these data, the vibrational temperature Tv behind the front of shock waves was measured at temperatures 4000-10,800 K in undiluted oxygen. Determination of Tv was based on the measurements of time histories of absorbance for two wavelengths behind the shock front and on the results of detail calculations of oxygen absorption spectrum. Solving the system of standard quasi-one-dimensional gas dynamics equations and using the measured vibrational temperature, the time evolution of oxygen concentration and other gas parameters in each experiment were calculated. Based on these data, the oxygen dissociation rate constants were obtained for thermal equilibrium and thermal non-equilibrium conditions. Furthermore, the oxygen vibrational relaxation time was also determined at high temperatures. Using the experimental data, various theoretical and empirical models of high-temperature dissociation were tested, including the empirical model proposed in the present work.
DOIURLPMID [本文引用: 2]
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
[本文引用: 1]
DOIURLPMID [本文引用: 3]
The polarization singularities are directly generated by using plasmonic metasurfaces with the geometric phase profiles designed to form the Poincaré beams. Different morphologies of polarization topological structures of lemon, star, monstar, spiral, dipole and quadrupole are created by the superpositions of Laguerre-Gauss modes with different orders under orthogonal circular or linear polarization basis. The polarization ellipse patterns and topological features of the produced optical vector fields are analyzed to reveal the properties of the polarization singularities of C-points and L-lines, and the orbital angular momentum states are also measured. The demonstrated polarization singularities generated from the geometric metasurfaces will promise many potential applications related to optical polarization imaging, metrology, optical trapping and quantum information processing.
[本文引用: 2]
DOIURLPMID [本文引用: 1]
The spectroscopic Franck-Condon (FC) principle is extended to mechanochemistry. If the external force is applied rapidly (the sudden-force regime), then the transition between the potential energy surface and the force-modified potential energy surface is analogous to the optical electronic transition. Such a transition produces a nonequilibrium ensemble of vibrationally excited molecules. This excess of vibrational energy is another activation source in addition to the well-known reaction barrier modulation by the external force. In the same time, the nonequilibrium vibrational distribution implies nonstatistical kinetics of a mechanochemical transformation. Mechanochemical FC principle thus provides a conceptual picture for the sudden-force mechanochemistry and opens possibilities for quantitative calculations of the mechanochemical rates and mechanisms. Here we use it to compute the dissociation rates of a model diatomic molecule and to explain the selectivity in mechanochemical bond breaking in n-butane. The approach is predicted to be relevant for large-magnitude external forces, applied instantaneously. Otherwise, the excess vibrational energy will dissipate due to intramolecular vibrational redistribution and interaction with environment.
DOIURLPMID
In the case of nuclear incidents, radioiodine may be liberated. After incorporation it accumulates in the thyroid and by internal irradiation enhances the risk of cancer occurrence. By administering a large dose of non-radioactive iodine the uptake of radioiodine into the gland can be inhibited ("iodine blockade"). Biokinetic models using first order kinetics are not suited to simulate iodine blockade, as the uptake into the gland is mediated by a saturable active transport. Therefore, we integrated an uptake mechanism described by a Michaelis-Menten kinetic into a simple ICRP biokinetic model. We moreover added a total uptake blocking mechanism representing the Wolff-Chaikoff effect becoming active when the gland is saturated with iodine. The validity of the model was ascertained by comparison with IMBA software. The competition of radioiodine and stable iodine at the membrane carrier site was modeled according to the rate law for monomolecular reactions for competing substrates. Our simulations show that competition for the uptake at the membrane carrier site accounts for about 60% and the saturation of the gland with iodine for over 35% of the total protective efficacy that exceeds 95%. Following acute radioiodine exposure, it is preferable to administer a single large dose of stable iodine. In the case of continuous radioiodine exposure, a single dose of stable iodine is less effective than after an acute exposure and splitting the total available dose and shortening the dosage intervals enhance efficacy. Model-based simulations may be a useful tool to develop antidote dosage schemes for uncommon emergencies.
DOIURLPMID [本文引用: 6]
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
[本文引用: 3]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 7]
DOIURL [本文引用: 3]
针对大气层内高超声速飞行时的化学非平衡现象,建立了沿驻点线流动的空气中氧离解度的计算模型. 模型假设氮气在氧气未充分离解时不发生离解,并且不涉及边界层内的复合反应. 理论计算发现,空气中氧的离解度随飞行高度的增加呈先增后减的非单调变化规律,其原因是由于化学反应平衡移动与非平衡效应相互作用的结果. 这一结论得到了数值模拟结果的验证,同时也解释了文献中当飞行高度较高时真实气体效应减弱的现象. 基于驻点线的近似理论模型,计算得到了轴对称钝头体绕流流场中的最大氧离解度及边界层外缘温度随飞行速度和高度变化的等值线图谱,相关结果可以为工程设计所用.
DOIURL [本文引用: 3]
针对大气层内高超声速飞行时的化学非平衡现象,建立了沿驻点线流动的空气中氧离解度的计算模型. 模型假设氮气在氧气未充分离解时不发生离解,并且不涉及边界层内的复合反应. 理论计算发现,空气中氧的离解度随飞行高度的增加呈先增后减的非单调变化规律,其原因是由于化学反应平衡移动与非平衡效应相互作用的结果. 这一结论得到了数值模拟结果的验证,同时也解释了文献中当飞行高度较高时真实气体效应减弱的现象. 基于驻点线的近似理论模型,计算得到了轴对称钝头体绕流流场中的最大氧离解度及边界层外缘温度随飞行速度和高度变化的等值线图谱,相关结果可以为工程设计所用.
DOIURL [本文引用: 3]
DOIURL
DOIURLPMID [本文引用: 1]
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}