摘要/Abstract
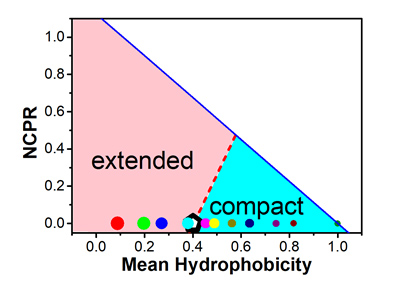
无序蛋白和折叠蛋白二者在结构和序列组成上存在着明显的差异.是疏水相互作用还是静电相互作用诱导了多肽结构的转变?在多肽结构转变过程中,疏水相互作用和静电相互作用各自发挥着什么样的作用?本工作以正(赖氨酸)、负(谷氨酸)和疏水性(异亮氨酸)的三种氨基酸为组分,产生了一系列电中性的无序随机多肽系统.利用全原子模型并采用蒙特卡洛方法进行了大规模计算模拟.结果表明,随着温度升高,多肽将从紧密构象转变到扩展构象.不同的多肽其转变温度依赖于疏水性氨基酸和带电氨基酸的比例.当平均疏水性低于临界疏水性时,转变温度低于室温;当平均疏水性大于临界疏水性时,转变温度高于室温.定量分析发现,临界疏水性数值与生物信息研究的结论是吻合的.此外,统计氨基酸残基之间的接触对数目表明,在多肽结构的转变过程中疏水作用发挥着主要作用.研究结果对蛋白质序列与结构关系的研究具有一定的理论指导意义,期望对基于序列的蛋白质全新设计提供参考.
关键词: 内禀无序蛋白, 构象, 序列, 疏水性, 蒙特卡洛模拟
Intrinsically disordered proteins (IDPs) are a unique class of proteins without stable native structures. Like globular proteins, the structure and the dynamics of IDPs are also encoded in their amino acid sequences. IDPs usually contain a larger proportion of hydrophilic or charged amino acids than globular proteins. Interestingly, even with the same hydrophobicity and number of charged residues, the differences in sequence arrangement can lead to different structures of the peptides. In this work, to model such an effect, we conduct molecular simulations based on a series of peptides with randomly composed of charged residues (including glutamic acids and lysines) and isoleucine. In the simulation, we use the ABSINTH (self-Assembly of Biomolecules Studied by an Implicit, Novel, and Tunable Hamiltonian) implicit solvation model and employ the all-atom Markov Chain Monte Carlo method with replica-exchange sampling. Our simulations clearly show a transition between the extended conformations to compact structures for each peptide. The corresponding transition temperature is found to be dependent on the portion of the hydrophobic and charged residues. When the mean hydrophobicity is larger than a certain threshold, the transition temperature is higher than the room temperature, and vice versa. Such a result has outlined the borderline between intrinsically disordered proteins and the folded proteins. It is also consistent with previous analysis based on bioinformatics techniques. Furthermore, the contributions of different kinds of interactions to the structural variation of peptides are analyzed based on the contact statistics and the charge-pattern dependence of the gyration radii of the peptides. Our simulation results imply that the hydrophobicity of the sequence dominates the order-disorder transitions of IDPs, while the charge distribution can also affect such transitions. Based on these results, we achieve a comprehensive understanding of the sequence-structure relation of the natural proteins and the underlying physics. Our results may broaden our perspective of the sequence-structure relation of protein systems and shed light on the design of both ordered and disordered proteins.
Key words: intrinsically disordered protein, conformation, sequence, hydrophobicity, Monte Carlo simulations
PDF全文下载地址:
点我下载PDF
