摘要/Abstract
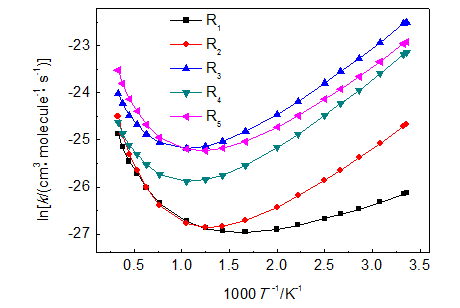
自由基与分子反应是一类具有负活化能的非基元反应,通常认为是通过反应复合物的两步过程,在大气化学和碳氢燃料燃烧机理中广泛存在,且在理论计算和实验上广泛研究.以碳氢燃料燃烧机理中重要反应类羟基自由基提取烷基过氧化氢α位氢的反应为研究对象,通过量化计算揭示其反应规律,计算得到其精确动力学参数.在所研究反应类中,定义第一步反应复合物的生成反应的标准摩尔吉布斯自由能变化等于零时所对应的温度为其转折温度Tc,并表明了当T >> Tc时可采用稳态近似法处理该类反应体系,得到总包反应速率常数.所有反应涉及的物种几何结构优化和频率分析均在BHandHLYP/6-311G(d,p)水平下得到,并在所研究反应类中选取了5个代表反应,通过CCSD(T)/CBS单点能计算,得到其最高转折温度为195.17 K,远远低于碳氢燃料燃烧模拟通常关注温度范围的最低温度650 K,表明用稳态近似法处理该类负活化能反应体系是合理的.计算还表明,该类反应的过渡态反应中心几何结构守恒,因此可将等键反应方法引入类反应,通过对低水平从头算得到的反应能垒进行校正,以得到高精度的结果.为了验证等键反应方法的可靠性,选取5个反应作为研究对象,将低水平BHandHLYP/6-311G(d,p)的校正结果和高水平CCSD(T)/CBS直接计算的结果进行比较,反应能垒最大绝对偏差由校正前的19.99 kJ·mol-1降到校正后的1.47 kJ·mol-1,表明用等键反应方法,只需在低水平从头算水平下就可以得到高水平的计算结果,从而可解决大分子体系精确动力学参数缺乏的问题.利用等键反应方法计算了20个反应的反应能垒,并结合过渡态理论计算得到了总包反应的速率常数,并揭示了该类反应只在低温段呈现负活化能关系.
关键词: 烷基过氧化氢氢提取, 反应复合物, 反应类等键反应方法, 速率常数, 负活化能
The reaction class of a free radical with a molecule are non-elementary reactions with negative activation energies and they are usually proceeded through two reaction steps with the first step being a reactant complex formation. This class of reactions are widespread in the atmospheric chemistry and the mechanism of hydrocarbon fuel combustion, so they are extensively studied in the theoretical calculation and experimental studies. The reaction class of α-H abstraction from alkyl hydroperoxides (ROOH) by hydroxyl radicals, which are important in the mechanism of hydrocarbon fuel combustion, are chosen as the object of this study. The regularity of this reaction class are revealed by quantum chemical calculations and their kinetic parameters are accurately calculated. When the standard molar Gibbs free energy change of the formation of the reactant complex in the first step is equal to zero, the corresponding temperature is defined as the conversion temperature Tc in this study, and it is shown that a steady state approximation method are applicable for this kind of reaction system to calculate the overall reaction rate constants when the temperature is much higher than the Tc. Geometric optimization and frequency analysis for all species were conducted at the BHandHLYP/6-311G(d,p) level. Five reactions are chosen as the representative for the reaction class and their single point energies are calculated using the method of CCSD(T)/CBS and it is shown that the highest conversion temperature for the five reactions is 195.17 K, far below usual modeling lowest temperature of the hydrocarbon fuel combustion, and therefore, the steady state approximation method is reasonable. It is also shown that the reaction-center geometries of the transition states are conserved, and thus the isodesmic reaction method is applicable to this reaction class to correct the energy barriers and rate constants at low-level BHandHLYP method. The obtained energy barriers are compared with the results using high-level ab initio CCSD(T)/CBS method and it is shown that the maximum absolute deviation of reaction energy barriers can be reduced from 19.99 kJ·mol-1 before correction to 1.47 kJ·mol-1 after correction, indicating that the isodesmic reaction method are applicable for the accurate calculation of the kinetic parameters for large-size molecular systems with the negative activation energy reaction. Finally, energy barriers for 20 reactions in the class are calculated with the isodesmic reaction method, and then based on steady state approximation, the rate constants for the overall reactions are calculated using the transition state theory in combination with the isodesmic correction scheme. It is shown that the negative activation energy relationship for the reaction class only exists in the low temperature region.
Key words: hydrogen abstraction of alkyl hydroperoxides, reactant complex, reaction class isodesmic reaction method, rate constant, negative activation energy
PDF全文下载地址:
点我下载PDF
