摘要/Abstract
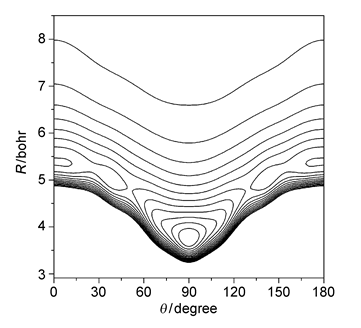
用高精度的量子化学从头算方法构建了Kr-CS2体系的精确的四维势能面.对该势能面不仅考虑了分子间的振动方式,而且考虑了单体CS2分子内的ν1对称伸缩和ν3反对称伸缩.采用了量子化学超分子方法在[CCSD(T)]-F12水平上构建了从头算势能面.将得到的四维势能面作积分,便得到CS2处于振动基态和激发态的复合物平均势能面,这两个态的势能面均有一个T型的全局最小值和两个相等的线性极小值.还通过径向部分采用离散变量表象法(DVR)和角度部分采用有限基组表象法(FBR)结合Lanczos循环算法对Kr-CS2复合物的振转能级进行了计算,对光谱常数进行了预测.另外,也预测了Kr-CS2复合物的带心位移(-1.2357 cm-1).
关键词: Kr-CS2, 从头算, 势能面, 红外光谱
The spectra of the van der Waals (vdW) complexes provide useful information on the intermolecular potential energy surfaces (PESs) and dynamics of such weakly bound molecules. First and foremost, an accurate potential energy surface is required to allow for spectroscopic analysis for van der Waals complexes. Thus, constructing an effective reduced-dimension potential energy surface, which includes direct relevant intramolecular modes, is the most feasible way and widely used in the recent potential studies. In this work, we present a four-dimensional (4D) ab initio potential energy surface (PES) of the Kr-CS2 complex at the coupled-cluster singles and doubles with noniterative inclusion of connected triples[CCSD(T)]-F12 level. We employed the aug-cc-pVTZ basis set of Woon and Dunning for the C and S atoms and the aug-cc-pVTZ-PP basis set for Kr. The bond functions (3s3p2d1f1g) (for 3s and 3p, α=0.9, 0.3, 0.1; for 2d, α=0.6, 0.2; for f and g, α=0.3) were placed at the mid-point of the R vector. The Q1 and Q3 normal modes for the ν1 symmetric stretching vibration and ν3 antisymmetric stretching vibration of CS2 were explicitly taken into account in the calculations of the Kr-CS2 potential energies. Two vibrationally averaged potentials with CS2 at both the vibrational ground and the ν1+ν3 excited states were generated from the integration of the four-dimensional potential over the Q1 and Q3 coordinates. Each potential contains a T-shaped global minimum and two equivalent linear local minima. These fits to 9000 points have root-mean-square (rms) deviations of 0.143 and 0.145 cm-1 for the ground and the ν1+ν3 excited states, respectively. The radial discrete variable representation (DVR)/angular finite basis representation (FBR) method and Lanczos algorithm were employed to calculate the rovibrational states without separating the inter-and intra-molecular vibrations. The spectroscopic parameters for the ground and the ν1+ν3 excited states of Kr-CS2 are predicted. In addition, the predicted band origin shift is -1.2357 cm-1 for Kr-CS2.
Key words: Kr-CS2, ab initio, potential energy surface, infrared spectra
PDF全文下载地址:
点我下载PDF
