摘要/Abstract
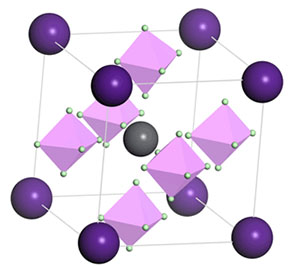
卤化物钙钛矿(ABC3)太阳能电池由于其优异的光电性质引起了人们的广泛关注,对结构中的A位及B位离子进行等价离子取代是调控钙钛矿材料禁带宽度的有效方法之一.本文研究拟用超卤素离子团簇(BeX3-,MgX3-,BX4-,AlX4-,SiX5-,PX6-,X=F,Cl)对立方相CsPbI3钙钛矿中的I-离子进行部分掺杂或完全取代,设计了一系列基于超卤素团簇掺杂的新型卤化物钙钛矿太阳能电池材料,并运用第一性原理的方法对材料的结构和性质进行了模拟研究.结果表明,采用上述超卤素离子团簇对CsPbI3钙钛矿中的I-离子进行部分掺杂比全部取代后得到的结构体系禁带宽度小;采用PCl6-团簇取代I-离子得到的CsPb (PCl6)3体系的禁带宽度是1.58 eV,其导带底部主要由Cl原子的3p轨道、P原子的3s轨道以及Pb原子的6p轨道杂化组成,该材料是一种潜在的钙钛矿太阳能电池材料.
关键词: 卤化物钙钛矿, 太阳能电池, 超卤素, 掺杂, 第一性原理
Halide perovskite (ABC3) solar cell has received a lot of attentions due to its excellent photoelectronic properties. It has been proven to be an effective way to modify halide perovskite materials' bandgap by replacing A or B ions with other equivalent ions. However, C ions have much fewer choices and are limited to halogen anions or pseudohalides anions. We designed a series of new cubic perovskite structures through substituting C anions by superhalogen clusters anions (BeX3-, MgX3-, BX4-, AlX4-, SiX5-, PX6-, X=F, Cl), and studied their structures and properties in first-principles way. Calculations were performed by using the Vienna ab initio simulation package (VASP) based on density functional theory. The DOS (Density of States) and bandgaps were calculated to analyze properties of the new perovskite structures. The results show that BeX3-, MgX3- (X=F, Cl) and SiCl5- could not remain its structure which means these three clusters are not superhalogen anions anymore after doping. The size and symmetry of superhalogen anions have influences on the structures of doped perovskites. The superhalogen anion whose symmetry is higher and size is closed to I- ion induces less distortions to doped perovskite structures. Comparing to the VBM (Valence Band Maximum) and CBM (Conduction Band Minimum) of CsPbI3, superhalogen anions substitutions could change the compositions of CBM and VBM and bandgaps. The bandgaps of superhalogen anions partial substitutions in halide perovskite become smaller compared to structures with superhalogen anions substituting completely. We demonstrate that the CsPb(PCl6)3, with a direct-bandgap of 1.58 eV located at M(0,0.5,0.5) point, could be a potential candidate material for solar cells. Its CBM mainly is dominated by Cl 3p states, P 3s states and Pb 6p states. The other doped perovskites with wide bandgaps may have potential applications in transistors or memristors. We hope that these results could provide theoretical guidance for synthesis of new perovskite materials for solar cells.
Key words: halide perovskite, solar cell, superhalogen, substitutions, first-principles
PDF全文下载地址:
点我下载PDF
