 , 赖伟斌2, 曾宇飞1, 卢桂宁1, 党志1, 郭楚玲1
, 赖伟斌2, 曾宇飞1, 卢桂宁1, 党志1, 郭楚玲1
1. 华南理工大学环境与能源学院, 工业聚集区污染控制与生态修复教育部重点实验室, 广州 510006;
2. 仲恺农业工程学院环境科学与工程学院, 广州 510225
收稿日期: 2020-03-12; 修回日期: 2020-04-10; 录用日期: 2020-04-10
基金项目: 国家自然科学基金(No.41977277,41720104004);广东省自然科学基金(No.2018A030313918)
作者简介: 姜梦戈(1995-), 女, E-mail:201720142517@mail.scut.edu.cn
通讯作者(责任作者): 郭楚玲, E-mail:clguo@scut.edu.cn
摘要:铁硫酸盐次生矿物广泛存在于酸性矿山废水(AMD)污染流域沉积物中,是多种有毒有害重金属的沉淀库.硫酸盐还原菌对矿物的还原作用可破坏矿物的稳定性从而引起共沉淀重金属的释放.在前期研究中发现柠檬酸杆菌Citrobacter sp.存在于AMD流域沉积物中,且在还原条件下具有较强的硫酸盐还原能力.因此,探究其对铁硫次生矿物的还原溶解及矿物晶相转化行为的影响尤为重要.本研究选取矿区沉积物中典型的铁硫次生矿物施氏矿物和黄钾铁矾为研究对象,在实验室条件下考察了从AMD污染沉积物中筛选到的菌株Citrobacter sp.EBS8对矿物的还原溶解及相转化的影响.结果表明,在缺氧、中性pH及电子供体足量时,在EBS8的作用下施氏矿物和黄钾铁矾均出现了明显的还原溶解现象.菌株EBS8可利用乳酸盐作为电子供体还原SO42-和Fe(III).SEM结果显示,施氏矿物在反应初期出现表面毛刺不平现象,黄钾铁矾发生由外向内的逐层溶解,矿物转化过程中都出现空壳形貌.XRD检测到施氏矿物组的主要转化产物为菱铁矿和蓝铁矿,黄钾铁矾体系中主要是蓝铁矿和马基诺矿.这些结果有助于了解AMD污染沉积物中铁硫酸盐矿物的转化行为及为AMD污染治理提供理论支撑.
关键词:铁硫酸盐次生矿物Citrobacter sp.还原溶解相转化
Effects of Citrobacter sp. EBS8 on reductive dissolution and phase transformation behaviors of schwertmannite and jarosite
JIANG Mengge1
, LAI Weibin2, ZENG Yufei1, LU Guining1, DANG Zhi1, GUO Chuling11. The Key Laboratory of Pollution Control and Ecosystem Restoration in Industrial Clusters, Ministry of Education, School of Environment and Energy, South China University of Technology, Guangzhou 510006;
2. School of Environmental Science and Engineering, Zhongkai University of Agriculture and Engineering, Guangzhou 510225
Received 12 March 2020; received in revised from 10 April 2020; accepted 10 April 2020
Abstract: Fe(Ⅲ)-oxyhydroxysulfate minerals are ubiquitous in sediments in rivers contaminated by acid mine drainage (AMD), and considered as scavengers for heavy metals. The reduction of these minerals by sulfate reducing bacteria destroys the stability of minerals and causes the release of coprecipitated contaminants. Previous researches had found Citrobacter sp. existed in the AMD sediment with a high sulfate reduction ability under reducing condition. Therefore, it is crucial to explore the reduction and dissolution of Fe(Ⅲ)-oxyhydroxysulfate minerals and the transformation behavior of mineral crystalline phase by Citrobacter sp.. In this study, schwertmannite and jarosite, the typical Fe(Ⅲ)-oxyhydroxysulfate minerals, were selected to investigate the effect of Citrobacter sp. EBS8 on the reductive dissolution and mineral phase transformation under laboratory conditions. The results showed that both schwertmannite and jarosite dissolved and had phase transformation in anoxic, neutral and sufficient electron donor systems. Strain EBS8 could reduce SO42- and Fe(Ⅲ) by using lactate as electron donor. Scanning electron micrograph (SEM) further showed that the surface burr of schwertmannite appeared in the initial phase, and jarosite was dissolved layer by layer from the outside to the inside, and then appeared morphology of the empty shell. The main transformation products of schwertmannite were siderite and vivianite, while jarosite was transformed into vivianite and mackinawite. The present study helps to understand the transformation behavior of Fe(Ⅲ)-oxyhydroxysulfate minerals and provide theoretical support for remediation of AMD-contaminated environment.
Keywords: Fe(Ⅲ)-oxyhydroxysulfate mineralsCitrobacter sp.reductiondissolutionphase transformation
1 引言(Introduction)酸性矿山废水(Acid Mine Drainage, AMD)的低pH、高SO42-含量和富含高铁的环境, 极易形成施氏矿物(Schwertmannite, Fe8O8(OH)6SO4)和黄铁矾类(Jarosite, K, Na, NH4, H3O[Fe3(OH)6(SO4)2])等铁硫酸盐次生矿物(Equeenuddin et al., 2013), 同时伴随着多种重金属(如铜、锌、镉、铬等)的存在(Xie et al., 2018).由于黄铁矾类及施氏矿物在成矿过程中, 矿物晶格结构中的多个位点易被其他性质相似的离子所取代, 如施氏矿物中的Fe3+可被Cu2+、Pb2+、Cd2+等阳离子取代, SO42-则有可能被离子半径及电荷相当的CrO42-、AsO43-、MoO43-等含氧阴离子取代, 从而使这些有毒有害物质进入矿物相而被固定(Regenspurg et al., 2005; Fiol et al., 2013).因此, 这类次生矿物被认为是AMD流域中重要的沉淀库(周立祥, 2008).
铁硫酸盐次生矿物的稳定性差, 受环境扰动易发生溶解和相转化, 从而影响共沉淀物质的归趋并造成潜在的环境风险(Regenspurg et al., 2003).如温度升高至80 ℃会使施氏矿物向赤铁矿转变(Regenspurg et al., 2005);随着pH的升高黄钾铁矾依次转化为针铁矿、赤铁矿、水铁矿(Madden et al., 2012).Burton等(2008)研究发现, 施氏矿物的溶解和相转化与微生物的铁硫还原相关.在异化铁还原菌Shewanella putrefaciens的还原作用下, 负载Pb的黄铁矾类矿物溶解转化形成二次矿物(Smeston et al., 2009).硫酸盐还原菌(Sulfate Reduction Bacteria, SRB)不仅可以利用硫酸盐作为电子受体, 也可直接或间接还原Fe(Ⅲ) (Sánchez-Andrea et al., 2014), 从而影响铁硫次生矿物结构的稳定性.Li等(2006)发现Desulfovibrio desulfuricans G-20可以在无SO42-的情况下, 直接通过酶还原固态三价铁.此外, 在硫酸盐和铁氧化物共存体系中, 硫酸盐还原产物S2-可将矿物中的固态Fe(Ⅲ)还原为Fe2+, 而自身被氧化为S0, 并在SRB的作用下再次被还原, 形成以硫单质为电子传递体的循环路径并驱动铁还原的发生(Hansel et al., 2015).
肠杆菌科柠檬酸杆菌属(Citrobacter)中的多个菌株已被报道具有硫酸盐还原能力.Qiu等(2009)从大宝山矿区分离出的Citrobacter sp. DBM菌株属于以硫酸盐为硫源的非传统SRB, 能在好氧条件下快速生长, 转至厌氧条件可在7 d内将10 mmol·L-1的硫酸盐完全还原为硫化物.Zhang等(2017)在处理AMD的上流式厌氧污泥反应床污泥中分离出一株类似菌株Citrobacter freundii Sr 10, 其对重金属Tl及SO42-的去除率分别可达到99%和89%.本课题组前期在大宝山矿区AMD污染流域多个不同pH值的沉积物中均检测到Citrobacter sp.的存在, 值得注意的是, 在AMD源头的极端酸性环境中(pH < 3.0), Citrobacter在属分类水平上仍然有较高的相对丰度(Bao et al., 2016).Zeng等(2018)采用土著SRB群落间接接触作用于施氏矿物时, Citrobacter sp.在群落中占优势;Gao等(2019)在将SRB群落与黄钾铁矾共培养时, 也发现类似现象.目前已报道的关于柠檬酸杆菌属的硫酸盐还原菌的研究更多地关注于其对重金属和SO42-的去除能力, 但对铁硫次生矿物稳定性的影响尚不清楚, 因此, 有必要进一步厘清在还原条件下Citrobacter sp.对铁硫酸盐次生矿物的作用.本研究选取铁硫矿区AMD污染流域沉积物中典型的铁硫次生矿物(施氏矿物和黄钾铁矾)为研究对象, 考察从AMD污染流域沉积物中筛选到的菌株Citrobacter sp. EBS8对矿物的还原溶解及相转化的影响, 以期为AMD污染环境中铁硫元素地球化学过程及相关污染物的迁移转化行为研究提供理论依据.
2 材料与方法(Materials and methods)2.1 菌株培养实验采用从广东省大宝山矿区AMD污染流域(横石河流域)下游沉积物中分离筛选得到的具有硫酸盐还原能力的柠檬酸杆菌(Citrobacter sp. EBS8), 其16S rRNA测序序列在美国国家生物技术信息中心(National Center for technology Information, NCBI)中归档的登陆号为MN 420979.
生长培养基为改良后的PostgateB培养基(Postgate, 1979), 配方如下:3.5 mmol·L-1 KCl、5 mmol·L-1 NH4Cl、0.5 mmol·L-1 KH2PO4、0.05 mmol·L-1 CaCl2、0.1 mmol·L-1 MgCl2、10 mmol·L-1 Na2SO4、30 mmol·L-1乳酸钠, 用1:1 HCl调节pH值至6.0, 121 ℃灭菌30 min后使用.批实验所用培养基为无添加Na2SO4的培养基.所用试剂均从阿拉丁试剂公司购买.
Citrobacter sp. EBS8接种在装有改良PostgateB培养基的厌氧瓶中, 置于30 ℃、150 r·min-1摇床中振荡扩培, 到达指数生长阶段后停止培养(约5~7 d).将培养液在4 ℃下10000 r·min-1离心10 min, 并用无菌水洗3次, 最后悬浮于无SO42-的无菌培养基中制备成OD600为0.2的细菌悬浮液.
2.2 矿物合成施氏矿物参考Regenspurg等(2003)的合成方法, 简述为:在2 L的洁净烧杯中添加988 mL超纯水、22.24 g FeSO4·7H2O, 用玻璃棒搅拌使试剂完全溶解;随后将12 mL H2O2(30%)缓慢滴加到溶液中, 并不断用玻璃棒搅拌, 滴加完后静置24 h;离心弃去上清液, 并用硫酸酸化至pH=2的超纯水洗涤3次, 再用超纯水洗涤若干次至上清液pH不变;离心后冷冻干燥, 研磨过200目筛, 避光干燥保存备用.
黄钾铁矾参考Baron等(1996)的合成方法, 简述为:在250 mL洁净烧杯中加入100 mL超纯水、17.2 g Fe2(SO4)3·5H2O、5.6 g KOH, 用玻璃棒搅拌使药剂完全溶解;将烧杯置于95 ℃恒温水浴锅中4 h, 并不断搅拌;静止冷却后离心弃去上清液, 并用超纯水洗涤3次;离心后冷冻干燥, 研磨过200目筛, 避光干燥保存备用.
矿物合成后, 采用扫描电镜(SEM, Carl Zeiss Microscopy, Germany)观察矿物形貌, 并采用X射线衍射光谱(XRD, D8 ADVANCE, Bruker)确定合成矿物种类.如图 1所示, 合成矿物的SEM图像、XRD谱图与已有文献报道的施氏矿物(Fan et al., 2019)和黄钾铁矾(Gao et al., 2019)一致.
图 1(Fig. 1)
 |
| 图 1 合成施氏矿物(SCH)与黄钾铁矾(J)的SEM图像(a.SCH, b.J)和XRD谱图(c.SCH, d.J) Fig. 1SEM images(a.SCH, b.J) and XRD spectrums(c.SCH, d.J) of synthetic schwertmannite and jarosite |
2.3 菌株EBS8与矿物共培养实验批实验分别对施氏矿物和黄钾铁矾两种矿物设置无菌对照组(SCH、J)、施氏矿物组(SCH+Bio)、黄钾铁矾组(J+Bio).批实验在120 mL的厌氧瓶中进行, 将180 ℃干热灭菌2 h后的矿物粉末以2 g·L-1的剂量添加至厌氧瓶中, 添加79 mL不含SO42-的改良PostgateB培养基和1 mL细菌悬浮液.摇匀, 用聚四氟乙烯橡胶塞密封、加铝盖, 置于30 ℃、150 r·min-1避光摇床中培养.每个处理设置3个平行, 定时取样, 悬浊液样品过0.22 μm滤膜后测定溶液的pH、Fe(Ⅱ)、SO42-、K+(黄钾铁矾组)、乳酸盐(Lactate)、乙酸盐(Acetate).另取悬浊液离心弃上清液, 冷冻干燥后得固体样品, 用于XRD、FTIR、SEM的表征.
2.4 分析方法与仪器本研究采用邻菲罗啉显色法并用紫外分光光度计(UV-160A, Shimadzu, Japan)在510 nm波长下测定溶液中Fe2+的浓度;用火焰原子吸收法测定溶液中K+浓度(Optima 5300DV Perkin-Elmer, San Diego, USA);小分子酸(Lactate和Acetate)在液相中的浓度采用高效液相色谱仪(HPLC, Angilent LC 1260, USA)测定(Xia et al., 2019);采用离子色谱仪(Dionex ICS-1500, USA)测定溶液中SO42-的浓度;固相SO42-含量的检测:精确称取定量固相样品, 在10 mL 0.05 mol·L-1的HCl和0.05 mol·L-1的HNO3溶液中加热至80 ℃, 持续1 h后重新定容(曾宇飞, 2018), 随后采用离子色谱仪检测.质量控制:采用标准样品控制分析方法的精确度, 使用的标准溶液有GSB04-1726-2004(铁)、GSB04-1733-2004(钾)及GSB04-1773-2004(硫酸根);采用3次平行样分析取平均值的方法控制指标分析的精密度, 并以3次分析的相对偏差作为分析误差.固相样品采用傅里叶变换红外光谱(FTIR, VERTEX 33, Bruker)对矿物表面官能团变化进行分析, 采用SEM观察矿物形貌变化, 采用XRD确定矿物相组成.
3 结果和讨论(Results and discussion)3.1 溶液理化因子变化经过80 d的培养周期, 两种矿物反应体系的pH值均发生了显著变化.如图 2所示, pH值在无菌对照组添加矿物后都迅速下降随后维持稳定, 施氏矿物和黄钾铁矾对照组的pH值分别从6.0下降至5.0和5.5.环境中施氏矿物及黄钾铁矾稳定存在的pH分别为2.5~4.5和 < 3 (Bao et al., 2017), 当矿物所处体系背景pH值高于稳定范围时, 矿物会发生溶解甚至转化, 并释放氢离子降低体系pH值.相反, 在有菌的实验体系中pH值都呈上升趋势, 并在反应30 d后基本稳定.柠檬酸杆菌利用乳酸还原硫酸根是不完全氧化反应, 生成乙酸和二氧化碳, 同时升高体系pH值(Zeng et al., 2019), 具体见式(1).最终, 施氏矿物和黄钾铁矾有菌组体系pH分别稳定至7.0和6.5左右.研究表明, 微生物消耗铁硫酸盐次生矿物中的Fe(Ⅲ)是升高体系pH的过程(Bao et al., 2017);另外, 矿物转化产物的形成对溶液体系的pH值有影响, 例如, 菱铁矿(FeCO3)(Zachara et al., 2002)和硫化亚铁(FeS)(Li et al., 2018)的产生均会减缓pH值的上升, 具体见式(2)~(3).
图 2(Fig. 2)
 |
| 图 2 溶液pH随反应时间的变化 (a.施氏矿物, b.黄钾铁帆) Fig. 2Changes of solution pH with time (a.Schwertmannite, b.jarosite) |
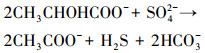 | (1) |
 | (2) |
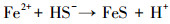 | (3) |
图 3(Fig. 3)
 |
| 图 3 施氏矿物组和黄钾铁矾组乳酸(a.SCH, c.J)与乙酸(b. SCH, d.J)的浓度变化 Fig. 3The variations of lactate (a.SCH, c.J) and acetate (b.SCH, d.J) in Schwertmannite and jarosite treatments |
两种矿物体系中溶解性SO42-、Fe2+及黄钾铁矾体系中溶解性K+浓度随时间的变化如图 4所示.由图可知, 无菌对照组中, 两种矿物添加到体系后均有SO42-的释放.原因可能是施氏矿物表面吸附的部分SO42-释放到溶液中(Paikaray et al., 2010), 且在pH值升高后, 羟基与矿物结合有利于结构中金属的脱落, 从而提高矿物的溶解转化(Xie et al., 2017).黄钾铁矾在中性环境下存在自发溶解现象(Madden et al., 2012), 导致结构中SO42-的释放, 对照组溶液中少量K+的检出也证实了这一结果(图 4e).在有菌体系中, 细菌生长引起pH值升高, SO42-快速释放, 在第3 d达到峰值后下降, 随后两组有菌体系中SO42-的含量下降到几乎为零(图 4a和4c);同时, 黄钾铁矾组(J+Bio)中K+浓度持续上升, 然而溶液中的SO42-和Fe2+浓度并未升高, 表明黄钾铁矾溶解释放的铁和硫可能再次沉淀为固相.在硫酸盐还原菌的作用下, 硫酸盐矿物释放出的SO42-随即被还原, 这是一个动态的还原溶解过程(Gao et al., 2019).且SO42-被还原后产生的S2-有能力还原溶解性Fe3+和攻击矿物结构中的Fe(Ⅲ)(He et al., 2019), 造成Fe2+的积累, 具体见式(4).从图 4b和4d可以看出, 溶液中Fe2+的积累滞后于SO42-的还原, 表明在EBS8的还原作用下, 硫酸根比三价铁更易得到电子.已有研究表明, 尽管Fe3+还原所需的能量及电子少于SO42-还原, 但硫酸盐还原菌更倾向于利用SO42-作为电子受体而非Fe3+(Hansel et al., 2015), 且高结晶度的铁氧化物更有利于硫还原的发生.总Fe浓度变化(未展示)与Fe2+浓度变化一致, 说明体系中并未检测到可溶性Fe3+.在高pH条件下溶解性的Fe3+可通过水解或沉淀作用快速吸附到固相表面(Kendall et al., 2013), 故未检测到矿物溶解释放的Fe3+.
图 4(Fig. 4)
 |
| 图 4 施氏矿物组和黄钾铁矾组中溶解性SO42-(a. SCH, c. J)、Fe2+(b. SCH, d. J)和黄钾铁矾体系中溶解性K+(e)浓度随时间的变化 Fig. 4Variation curves of SO42-(a. SCH, c. J), Fe2+(b. SCH, d. J) in experimental solutions in each systems and K+ (e) in the systems of jarosite |
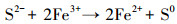 | (4) |
反应至80 d时, 反应体系中剩余SO42-在液相和固相中的含量如表 1所示.施氏矿物空白对照组中SO42-的总含量为2.62 mmol·g-1, 而在有菌实验体系中, 仅在剩余固相产物中检测到1.13 mmol·g-1, 表明在菌株EBS8的还原作用下促进了矿物的溶解, 并将总SO42-还原56.9%左右.同样地, 在黄钾铁矾有菌实验体系中SO42-的剩余量为1.05 mmol·g-1, EBS8对黄钾铁矾中SO42-的还原溶解率为60.8%左右.猜测有菌体系固相中剩余SO42-由于矿物的相转化作用合成到新矿物内或吸附在新矿物表面, 新矿物形成覆盖层对剩余矿物的还原溶解产生阻碍作用(Fredrickson et al., 2003), 导致SO42-的还原不完全, 因此, 需要对矿物的转化产物进一步检测.
表 1(Table 1)
| 表 1 第80 d时各处理体系中SO42-的含量分布 Table 1 Contents distribution of SO42- in systems at the 80th day | ||||||||||||||||||||
表 1 第80 d时各处理体系中SO42-的含量分布 Table 1 Contents distribution of SO42- in systems at the 80th day
| ||||||||||||||||||||
3.2 固体矿物学分析3.2.1 FTIR分析施氏矿物及黄钾铁矾的FTIR谱图如图 5所示.初始施氏矿物(图 5a)在418 cm-1和701 cm-1处的吸收峰对应矿物结构中Fe—O伸缩振动;611 cm-1处为矿物隧道结构内的SO42-分子振动v4(SO4), 979 cm-1处为SO42-分子内对称收缩振动的吸收峰v1(SO4), 被认为是施氏矿物外层络合的SO42-(Fan et al., 2019);1130 cm-1处为SO42-分子的三重简并反对称伸缩振动的吸收峰v3(SO4), 并有两个肩峰(1195 cm-1和1050 cm-1)存在(李君菲等, 2018);位于1630 cm-1的吸收峰是由H—O—H的伸缩振动产生的.无菌对照组中第80 d时固相产物的FTIR图中各吸收峰的峰强有微弱变化, 但峰位置及数量与原矿基本相同.在施氏矿物有菌实验体系中, 与Fe—O和SO42-有关的吸收峰强度逐渐减弱, 直至消失, 说明在微生物的作用下施氏矿物结构逐步瓦解至完全溶解.同时, 在858 cm-1和1013 cm-1处有新的吸收峰出现, 且峰强随反应时间逐渐加强.有文献报道, 858 cm-1和1013 cm-1处的吸收峰分别是PO43-(Gao et al., 2019)和S—S (Chernyshova, 2004)引起的, 猜测新形成的矿物中有蓝铁矿和黄铁矿的存在.在790 cm-1处出现新的吸收峰, 有文献指出此处为针铁矿的特征峰(王小明, 2015).在厌氧体系中, 生物还原所产生的硫化物能与重金属结合并反应生成更稳定的矿物质, 如黄铁矿可由硫化亚铁转化得到(Li et al., 2006).针铁矿的形成主要是由于Fe2+的存在导致矿物中Fe3+的释放, 解析出的Fe3+快速水解沉淀下来(Bigham et al., 1996), 这也是本研究过程中未检测到溶解性Fe3+的原因之一.在不同反应时间下黄钾铁矾的FTIR谱图中(图 5b), 573、509和475 cm-1处的峰指的是FeO6配位八面体的振动吸收峰;627、1086和1182 cm-1分别代表黄钾铁矾中SO42-的吸收峰v3(SO4)和v4(SO4);结构中O—H形变引起的吸收峰位置在1003 cm-1处;羟基质子化引起的H—O—H变形在1638 cm-1处产生微弱的吸收峰.随着反应时间的进行, SO42-和FeO6的峰强度减弱直至消失, 确认了反应过程中黄钾铁矾的结构破损和完全溶解.在1800~1200 cm-1之间吸收峰的变化多是由羧基和氨基引起的, 表明矿物转化与微生物活动有关(Yeasmin et al., 2014).尽管两种矿物在反应至80 d时都发生了溶解转化, 但其红外谱图有较大差异, 为进一步确认矿物是否在微生物的作用下结构破损直至完全溶解, 需对矿物的形貌进行观察.
图 5(Fig. 5)
 |
| 图 5 施氏矿物组(a)和黄钾铁矾组(b)在不同反应时间下矿物转化产物的FTIR图 Fig. 5FTIR spectra of samples at different time in Schwertmannite(a) and jarosite(b) treatments |
3.2.2 SEM分析矿物SEM图像(图 6和图 7)显示, 在两种无菌对照组中, 矿物的形貌和结构始终保持圆球状, 其中, 黄钾铁矾表面观察到少量点蚀现象, 与黄钾铁矾的自发溶解相对应, 可见施氏矿物和黄钾铁矾在本研究的空白培养基中培养80 d未发生明显溶解和相转化.在有菌体系中, 施氏矿物组(SCH+Bio)在反应至第15 d时, 矿物表面已经出现毛刺不平的现象, 随着反应的进行, 表面光滑的球状矿物越来越少, 并逐渐出现空壳结构和菱形块状矿物;反应至80 d时, 出现了体积更大的长条状矿物, 与已有文献报道中的蓝铁矿形貌类似(Wang et al., 2019).在黄钾铁矾组中(J+Bio)可观察到矿物的球体结构是由外到内逐层瓦解的, 已有研究利用缩核模型较好地模拟了黄钾铁矾的溶解过程(Ntumba Malenga et al., 2015).而后生物还原生成的S2-和Fe2+快速结合生成硫化物沉淀, 以原始矿物为附着点生长, 并以潜在的球状结构为底发育聚集, 与已有研究中的马基诺矿形貌相同(Johnston et al., 2012), 原始矿物逐层溶解后, 次生矿物形成空壳结构.
图 6(Fig. 6)
 |
| 图 6 施氏矿物反应体系中不同反应时间下矿物转化产物的SEM图 Fig. 6SEM images of samples at different time in Schwertmannite systems |
图 7(Fig. 7)
 |
| 图 7 黄钾铁矾反应体系中不同反应时间下矿物转化产物的SEM图 Fig. 7SEM images of samples at different time in jarosite systems |
3.2.3 XRD分析进一步用XRD对反应产物种类进行分析(图 8), 施氏矿物和黄钾铁矾两种矿物体系的无菌对照组中, 矿物在反应至80 d依然保留着原始矿物的特征峰, 表明尽管矿物的红外峰值有所减弱, 但发生相转化的矿物量很少.施氏矿物有菌实验组(SCH+Bio)在30 d时施氏矿物的特征峰完全消失, 并出现微弱的菱铁矿特征峰, 反应至80 d时, 菱铁矿特征峰逐渐增强并占主导, 蓝铁矿特征峰相对较弱, 与SEM的结果一致.施氏矿物组谱图中出现水磷三铁矿(Fe3+Fe23+(PO4)2(OH)3·5H2O)的特征峰, 是Fe3+与PO43-生成的水合矿物.有文献表明, 在还原条件下, 溶液中的PO43-可以将施氏矿物中的SO42-置换出来, 也可以吸附在施氏矿物表面与Fe3+形成红磷铁矿(FePO4)沉淀, 形成物理障碍, 从而增加了还原生成的二价铁与溶液中离子的距离, 更易生成菱铁矿(Zachara et al., 2002).但由于本研究中所添加PO43-为0.5 mmol·L-1, 完全沉淀后矿物量 < 10%, 故难以用XRD检测到(Schoepfer et al., 2019).黄钾铁矾有菌实验组在第15 d已经检测出蓝铁矿特征峰, 在30 d后马基诺矿的峰逐渐成为主要特征峰.马基诺矿是无定型的硫化亚铁矿物, 有文献表明马基诺矿是黄钾铁矾硫化后的主要产物(Johnston et al., 2012).红外谱图中未出现明显FeS吸收峰的原因是Fe—S引起的红外吸收峰比较微弱(Gong et al., 2014).XRD谱图中硫单质和黄铁矿出现微弱峰值, 表明体系中硫化产物化学还原Fe3+的存在.
图 8(Fig. 8)
 |
| 图 8 施氏矿物(a)和黄钾铁矾(b)转化产物的XRD谱图 Fig. 8XRD patterns of the precipitation in Schwertmannite(a) and jarosite(b) systems |
5 结论(Conclusions)本文在实验室条件下探究了一株具有硫酸盐还原能力的柠檬酸杆菌EBS8对两种铁硫酸盐次生矿物还原溶解的影响.结果表明, 在缺氧、中性pH值及电子供体足量时, 施氏矿物和黄钾铁矾在柠檬酸杆菌Citrobacter sp. EBS8的作用下发生明显的还原溶解现象, 推测在实际环境中菌株EBS8的存在可能会对两种矿物的稳定性产生威胁.细菌以矿物中的SO42-和Fe3+作为电子受体生成铁硫还原产物.在微生物和化学作用的综合影响下, 铁硫还原产物与溶液中的离子发生共沉淀作用, 形成一系列的Fe(Ⅱ)和Fe(Ⅲ)矿物.施氏矿物组的主要转化产物为菱铁矿和蓝铁矿, 黄钾铁矾体系中主要是蓝铁矿和马基诺矿, 这与矿物结构中Fe3+的释放和还原速率有关.
参考文献
| Bao Y, Guo C, Lu G, et al. 2017. Role of microbial activity in Fe(Ⅲ) hydroxysulfate mineral transformations in an acid mine drainage-impacted site from the Dabaoshan Mine[J]. Science of the Total Environment, 616-617: 647-657. |
| Bao Y, Guo C, Wang H, et al. 2016. Fe-and S-metabolizing microbial communities dominate an AMD-contaminated river ecosystem and play important roles in Fe and S cycling[J]. Geomicrobiology Journal, 8(34): 695-705. |
| Baron D, Palmer C D. 1996. Solubility of jarosite at 4~35℃[J]. Geochimica et Cosmochimica Acta, 60(2): 185-195. DOI:10.1016/0016-7037(95)00392-4 |
| Bigham J, Schwertmann U, Traina S, et al. 1996. Schwertmannite and the chemical modeling of iron in acid sulfate waters[J]. Geochimica et Cosmochimica Acta, 12(60): 2111-2121. |
| Burton E D, Bush R T, Sullivan L A, et al. 2008. Schwertmannite transformation to goethite via the Fe(Ⅱ) pathway:Reaction rates and implications for iron-sulfide formation[J]. Geochimica Et Cosmochimica Acta, 72(18): 4564. |
| Chernyshova I V. 2004. Pyrite oxidation mechanism in aqueous solutions:An in situ FTIR study[J]. Russian Journal of Electrochemistry, 40(1): 69-77. |
| Equeenuddin S M, Tripathy S, Sahoo P K, et al. 2013. Metal behavior in sediment associated with acid mine drainage stream:Role of pH[J]. Journal of Geochemical Exploration, 124: 230-237. DOI:10.1016/j.gexplo.2012.10.010 |
| Fan C, Fan C, Guo C, et al. 2019. Transformation of cadmium-associated schwertmannite and subsequent element repartitioning behaviors[J]. Environmental Science and Pollution Research, 26(1): 617-627. |
| Fan C, Guo C, Zeng Y, et al. 2019. The behavior of chromium and arsenic associated with redox transformation of schwertmannite in AMD environment[J]. Chemosphere, 222: 945-953. |
| Fiol S, Gondar D, Pérez C, et al. 2013. Cu(II) incorporation to schwertmannite:Effect on stability and reactivity under AMD conditions[J]. Geochimica Et Cosmochimica Acta, 119: 149-163. DOI:10.1016/j.gca.2013.05.029 |
| Fredrickson J K, Kota S, Kukkadapu R K, et al. 2003. Influence of electron donor/acceptor concentrations on hydrous ferric oxide (HFO) bioreduction[J]. Biodegradation, 14(2): 91-103. DOI:10.1023/A:1024001207574 |
| Gao K, Jiang M, Guo C, et al. 2019. Reductive dissolution of jarosite by a sulfate reducing bacterial community:Secondary mineralization and microflora development[J]. Science of the Total Environment, 690: 1100-1109. |
| Gong Y, Liu Y, Xiong Z, et al. 2014. Immobilization of mercury by carboxymethyl cellulose stabilized iron sulfide nanoparticles:Reaction mechanisms and effects of stabilizer and water chemistry[J]. Environmental Science & Technology, 48(7): 3986-3994. |
| Han X, Schultz L, Zhang W, et al. 2016. Mineral formation during bacterial sulfate reduction in the presence of different electron donors and carbon sources[J]. Chemical Geology, 435: 49-59. |
| Hansel C M, Lentini C J, Tang Y, et al. 2015. Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments[J]. The ISME Journal, 9(11): 2400-2412. DOI:10.1038/ismej.2015.50 |
| He L Y, Xie L, Wang D J, et al. 2019. Elucidating the role of sulfide on the stability of ferrihydrite colloids under anoxic conditions[J]. Journal of Geochemical Exploration, 124(1): 230-237. |
| Johnston S G, Burton E D, Keene A F, et al. 2012. Arsenic mobilization and iron transformations during sulfidization of As(V)-bearing jarosite[J]. Chemical Geology, 334: 9-24. DOI:10.1016/j.chemgeo.2012.09.045 |
| Karimian N, Johnston S G, Burton E D. 2018. Antimony and arsenic partitioning during Fe2+-induced transformation of jarosite under acidic conditions[J]. Chemosphere, 195: 515-523. DOI:10.1016/j.chemosphere.2017.12.106 |
| Kendall M R, Madden A S, Elwood Madden M E, et al. 2013. Effects of arsenic incorporation on jarosite dissolution rates and reaction products[J]. Geochimica Et Cosmochimica Acta, 112: 192-207. DOI:10.1016/j.gca.2013.02.019 |
| 李君菲, 谢莹莹, 党志, 等. 2018. 酸性矿山废水中铜在含铬施氏矿物上的吸附及其对矿物溶解与相转变的影响[J]. 环境科学学报, 38(8): 186-193. |
| Li G X, Chen X P, Wang X N, et al. 2018. Sulfur redox cycling dependent abiotic ferrihydrite reduction by a desulfitobacterium hafniense[J]. Acs Earth & Space Chemistry, 2(5): 496-505. |
| Li Y, Vali H, Yang J, et al. 2006. Reduction of iron oxides enhanced by a sulfate-reducing bacterium and biogenic H2S[J]. Geomicrobiology Journal, 23(2): 103-117. DOI:10.1080/01490450500533965 |
| Liu C, Sreenivas K, Zachara J M, et al. 2001. Kinetic analysis of the bacterial reduction of goethite[J]. Environmental Science & Technology, 35(12): 2482-2490. |
| Liu X, Gao C, Ji D, et al. 2017. Survival of Escherichia coli O157:H7 in various soil particles:importance of the attached bacterial phenotype[J]. Biology and Fertility of Soils, 53(2): 209-219. DOI:10.1007/s00374-016-1172-y |
| Madden M E E, Madden A S, Rimstidt J D, et al. 2012. Jarosite dissolution rates and nanoscale mineralogy[J]. Geochimica Et Cosmochimica Acta, 91: 306-321. DOI:10.1016/j.gca.2012.05.001 |
| Ntumba Malenga E, Mulaba-Bafubiandi A F, Nheta W. 2015. Alkaline leaching of nickel bearing ammonium jarosite precipitate using KOH, NaOH and NH4OH in the presence of EDTA and Na2S[J]. Hydrometallurgy, 155: 69-78. DOI:10.1016/j.hydromet.2015.04.004 |
| Paikaray S, Peiffer S. 2010. Dissolution kinetics of sulfate from schwertmannite under variable pH conditions[J]. Mine Water & the Environment, 29(4): 263-269. |
| Postgate J R. 1979. The Sulphate-Reducing Bacteria[M]. Cambridge, United Kingdom: Cambridge University Press. |
| Qiu R, Zhao B, Liu J, et al. 2009. Sulfate reduction and copper precipitation by a Citrobacter sp.isolated from a mining area[J]. Journal of Hazardous Materials, 164(2/3): 1310-1315. |
| Regenspurg S, Brand A, Peiffer S. 2003. Formation and stability of schwertmannite in acidic mining lakes 1 1 Associate editor:C.M.Eggleston[J]. Geochimica et Cosmochimica Acta, 68(6): 1185-1197. |
| Regenspurg S, Peiffer S. 2005. Arsenate and chromate incorporation in schwertmannite[J]. Applied Geochemistry, 20(6): 1226-1239. DOI:10.1016/j.apgeochem.2004.12.002 |
| Sánchez-Andrea I, Sanz J L, Bijmans M F M, et al. 2014. Sulfate reduction at low pH to remediate acid mine drainage[J]. Journal of Hazardous Materials, 269: 98-109. DOI:10.1016/j.jhazmat.2013.12.032 |
| Schoepfer V A, Burton E D, Johnston S G, et al. 2019. Phosphate loading alters schwertmannite transformation rates and pathways during microbial reduction[J]. The Science of the Total Environment, 657: 770-780. DOI:10.1016/j.scitotenv.2018.12.082 |
| Smeston C M, Fryer B J, Weisener C G. 2009. Intracellular Precipitation of Pb by Shewanella putrefaciens CN32 during the Reductive Dissolution of Pb-Jarosite[J]. Environmental Science & Technology, 43(21): 8086-8091. |
| 王小明.2015.几种亚稳态铁氧化物的结构、形成转化及其表面物理化学特性[D].武汉: 华中农业大学 |
| Wang Q, Wei Z, Yi X, et al. 2019. Biogenic iron mineralization of polyferric sulfate by dissimilatory iron reducing bacteria:Effects of medium composition and electric field stimulation[J]. The Science of the Total Environment, 684: 466-475. DOI:10.1016/j.scitotenv.2019.05.322 |
| Xia D, Yi X, Lu Y, et al. 2019. Dissimilatory iron and sulfate reduction by native microbial communities using lactate and citrate as carbon sources and electron donors[J]. Ecotoxicology and Environmental Safety, 174: 524-531. |
| Xie Y, Guining L, Chengfang Y, et al. 2018. Mineralogical characteristics of sediments and heavy metal mobilization along a river watershed affected by acid mine drainage[J]. Plos One, 13(1): e190010. |
| Xie Y, Guining L, Han Y, et al. 2017. Role of dissolved organic matter in the release of chromium from schwertmannite:Kinetics, repartition, and mechanisms[J]. Journal of Environmental Quality, 46(5): 1088-1097. DOI:10.2134/jeq2017.03.0122 |
| Yeasmin S, Singh B, Kookana R S, et al. 2014. Influence of mineral characteristics on the retention of low molecular weight organic compounds:A batch sorption-desorption and ATR-FTIR study[J]. Journal of Colloid & Interface Science, 432: 246-257. |
| Zachara J M, Kukkadapu R K, Fredrickson J K, et al. 2002. Biomineralization of poorly crystalline Fe(Ⅲ) oxides by Dissimilatory Metal Reducing Bacteria (DMRB)[J]. Geomicrobiology Journal, 19(2): 179-207. DOI:10.1080/01490450252864271 |
| Zeng Q, Hao T, Robert M H, et al. 2019. Recent advances in dissimilatory sulfate reduction:From metabolic study to application[J]. Water Research, 150: 162-181. DOI:10.1016/j.watres.2018.11.018 |
| Zeng Y, Wang H, Guo C, et al. 2018. Schwertmannite transformation via direct or indirect electron transfer by a sulfate reducing enrichment culture[J]. Environmental Pollution, 242: 738-748. |
| Zhang H, Li M, Yang Z, et al. 2017. Isolation of a non-traditional sulfate reducing-bacteria Citrobacter freundii sp.and bioremoval of thallium and sulfate[J]. Ecological Engineering, 102: 397-403. DOI:10.1016/j.ecoleng.2017.02.049 |
| 曾宇飞.2018.硫酸盐还原菌在不同电子转移模式下介导施氏矿物的转化过程[D].广州: 华南理工大学 |
| 周立祥. 2008. 酸性矿山废水中生物成因次生高铁矿物的形成及环境工程意义[J]. 地学前缘, 15(6): 74-82. |
