 , 李歆1, 沈翔森2, 曾立民1,4, 黄晓锋3, 朱波3, 林理量3, 楼晟荣2
, 李歆1, 沈翔森2, 曾立民1,4, 黄晓锋3, 朱波3, 林理量3, 楼晟荣2
1. 北京大学环境科学与工程学院, 环境模拟与污染控制国家重点联合实验室, 北京 100871;
2. 上海市环境科学研究院, 国家环境保护城市大气复合污染成因与防治重点实验室, 上海 200233;
3. 北京大学深圳研究生院, 环境与能源学院, 城市人居环境科学与技术实验室, 深圳 518055;
4. 南京信息工程大学, 江苏省大气环境与装备技术协同创新中心, 南京 210044
收稿日期: 2019-03-20; 修回日期: 2019-05-16; 录用日期: 2019-05-16
基金项目: 国家重点研发计划项目(No.2017YFC0209400);自然科学基金青年基金(No.21407107)
作者简介: 刘硕英(1993-), 女, E-mail:liushuoying@pku.edu.cn
通讯作者(责任作者): 楼晟荣, E-mail:lousr@saes.sh.cn
摘要: 羟基自由基(·OH)总反应性(kOH)是大气中所有·OH反应物的浓度与其·OH反应速率常数乘积的总和,对kOH的直接测量有助于识别未知的·OH反应物种及提升·OH收支分析的准确度.因此,本研究建立了一套基于激光光解-激光诱导荧光技术的kOH在线测量系统(LP-LIF),利用紫外脉冲激光在流动管内光解臭氧产生·OH,采用激光诱导荧光技术实时测量其与采样进入流动管的活性气体反应而导致的·OH浓度衰减,通过对该衰减进行指数拟合得到采样气的kOH.经实验室测试,LP-LIF系统对kOH的测量灵敏度为1.2 s-1,时间分辨率5 min.应用该系统对2018年秋季深圳地区的大气kOH进行为期1个月的连续测量,结合同步观测的·OH反应物浓度数据发现,kOH观测值在10~30 s-1之间,主要来自一氧化碳(14%)、氮氧化物(26%)和一次排放的挥发性有机物(24%).此外,由未测量的·OH反应物贡献的kOH平均约23%,且在夜间和早晚高峰时段贡献较高,推测其主要来自溶剂涂料、石化工业及LPG机动车排放.
关键词:OH自由基总反应性激光诱导荧光活性缺失
Measurement and partition analysis of atmospheric OH reactivity in autumn in Shenzhen
LIU Shuoying1,3
, LI Xin1, SHEN Xiangsen2, ZENG Limin1,4, HUANG Xiaofeng3, ZHU Bo3, LIN Liliang3, LOU Shengrong2 1. State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, Beijing 100871;
2. State Environmental Protection Key Laboratory of Formation and Prevention of the Urban Air Complex, Shanghai Academy of Environmental Sciences, Shanghai 200233;
3. Key Laboratory for Urban Habitat Environmental Science and Technology, School of Environment and Energy, Peking University Shenzhen Graduate School, Shenzhen 518055;
4. Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, Nanjing University of Information Science&Technology, Nanjing 210044
Received 20 March 2019; received in revised from 16 May 2019; accepted 16 May 2019
Abstract: The reactivity of hydroxy radical (·OH), namely kOH, is the sum of products between the reactant concentrations and their reaction rate constants with OH for all·OH reactants in the atmosphere. Direct measurements on kOH is helpful for identifying unknown·OH reactant and for improving the accuracy of·OH budget analysis. In this study, we developed an online kOH measurement system (LP-LIF) based on laser flash photolysis-laser induced fluorescence technique. The system uses a pulsed UV laser photolyzing ozone to generate·OH radical in a flow tube. By using the laser induced fluorescence technique to record the·OH concentration decay which is caused by the reaction of·OH with the sampled reactive gases in the flow tube, and by fitting the decay curve with an exponential function, the kOH of the sample gas can be derived. The measurement sensitivity at 5 min time resolution is 1.2 s-1, determined from laboratory test experiments. The LP-LIF system was applied in a 1-month field observation campaign in Shenzhen in autumn 2018. Simultaneous measurements were performed for·OH reactants including carbon monoxide (CO), nitrogen oxides (NOx), volatile organic compounds (VOCs), etc.. The observed kOH by LP-LIF is between 10 to 30 s-1, mainly coming from CO (14%), NOx (26%), and primarily emitted VOCs (24%). Moreover, it is found that on average around 23% kOH is from unmeasured·OH reactants. Since the contribution becomes higher during night and during morning and evening rush hours, we speculate that the missing kOH is originated from emissions of solvents, paints, petrochemical industry, and LPG vehicles.
Keywords: ·OH reactivitylaser-induced fluorescencemissing reactivity
1 引言(Introduction)OH自由基作为对流层大气中最重要的氧化物质, 是对流层大气化学的主要推动者(Ehhalt, 1999).它控制着大多数一次污染物和温室气体的寿命, 如甲烷、CO、挥发性有机物(VOCs)、NO2和SO2, 同时也导致了臭氧、硫酸和二次有机气溶胶(SOA)等二次污染物的生成(Stone et al., 2012).因此, 明确OH自由基的源和汇成为了理解对流层大气反应化学机制的关键所在.目前, OH自由基的来源较为明确, 主要包括O3、HONO和甲醛的光解, 烯烃和臭氧的反应, 以及过氧自由基在低NOx条件下的循环再生(Dolgorouky et al., 2012).但由于OH自由基的高活性与高反应性, 导致其反应物种类繁多、数量巨大, 而其去除渠道也相对复杂.对流层中大约存在104~105种VOCs(Goldstein et al., 2007), 但目前能够精确测量的仅百余种, 这便使OH自由基的去除方式评估产生了偏差.因此, 发展一种可以直接测量OH自由基总反应性的技术有助于解决这一问题, 从而加深人们对边界层大气光化学反应的理解.
OH自由基总反应性(kOH)是对大气中能与OH自由基反应的活性物质浓度的综合评价, 即大气中所有能与OH自由基反应的物质Xi的浓度同其与OH自由基反应速率常数(ki+OH)的乘积之和:
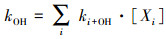 | (1) |
目前, 应用较为广泛的总反应性测量技术主要分为3类:第一类为直接测量法, 即通过测量人为产生的OH自由基随时间的衰减速率直接获得OH自由基总反应性, 这类方法包括移动注射-激光诱导荧光法(TOHLM, Total OH Loss-rate Measurement) (Kovacs et al., 2001)和闪光光解-激光诱导荧光法(LP-LIF, Laser flash Photolysis -Laser Induced Fluorescence) (Calpini et al., 1999);第二类为间接测量法, 即通过对比环境大气中活性物质和参比物种(如吡咯等)与OH自由基反应的相对竞争关系间接得到总反应性, 通常所用的方法为相对反应活性法(CRM, Comparative Reactivity Method) (Sinha et al., 2008);第三类为半直接测量法, 即通过测量其他物质的浓度得到OH自由基浓度, 继而通过OH自由基浓度随时间的衰减得到OH自由基衰减速率, 通常所用的方法为化学离子质谱法(CIMS, Chemical Ionisation Mass Spectrometry) (Muller et al., 2018).综上, 鉴于闪光光解-激光诱导荧光法(LP-LIF)与其他3种方法相比, 采用了臭氧光解的方法来生成OH自由基, 从而避免了HOx循环所带来的干扰, 在检测时该方法直接测量了OH自由基的衰减曲线, 并非通过OH自由基的浓度变化来间接计算得到总反应性值, 同时综合考虑时间分辨率、检出限与不确定性等因素, 该方法也都具有一定的优势, 因此, 闪光光解-激光诱导荧光法(LP-LIF)所获得的测量结果更为直接准确.本研究旨在采用此种方法搭建一套独立测量大气中OH自由基总反应性的装置, 并对深圳市秋季大气中的总反应性水平进行测量与分析.
2 外场观测与测量方法(Field measurements and methods)2.1 观测点位与时间深圳市地处我国东南部, 珠江三角洲东岸, 属于亚热带季风气候.观测地点位于深圳市南山区北京大学深圳研究生院内, 其位置为北纬22.59°, 东经113.98°, 周边有大型水库和疗养基地, 植被覆盖率较高, 附近以居民住宅区为主, 人口稠密, 交通系统发达, 邻近西丽镇商业区, 无明显局地污染源, 可在一定程度上代表深圳市大气污染的区域性特征.采样口架设于研究生院内C栋教学楼楼顶, 距地面约20 m.
测量时段为2018年10月1—18日, 其中, OH自由基总反应性的测量结果有3处数据缺失, 10月5—6日是由于仪器发生故障导致数据缺失, 10月10日及10月15—16日则是由于降雨的影响导致无法测量.
2.2 OH自由基反应活性测量方法本研究中OH自由基总反应性的测定装置主要结合了激光光解-激光诱导荧光法来完成测量.简单来讲, 就是将环境大气采入流动管后, 用波长为266 nm的高能量激光脉冲对其中的臭氧进行光解产生一定量的OH自由基, 使其在流动管内发生衰减, 进而借助激光诱导荧光(LIF)检测OH自由基的技术来记录这一衰减过程.
图 1为OH自由基总反应性测量装置的原理示意图, 图中的圆柱形框架代表了铝制流动管, 其内径为40 mm, 内部箭头所指代表了波长为266 nm的脉冲激光, 光束直径被扩束为30 mm后从流动管的中轴线处平行穿过.混合着臭氧和水的环境空气被匀速采入流动管时, 其速度要精确控制以保证OH自由基在管中的损失可测.流动管右下方采样嘴的流量约为流动管流量的15%.根据反应式(2)和(3)可知, 流动管内OH自由基的浓度仅与臭氧浓度、激光辐射强度和水分子浓度3个因素有关, 当发生一次激光光解时, 可认为辐射范围内的OH自由基浓度是一致的.
图 1(Fig. 1)
 |
| 图 1 流动管气流示意图 Fig. 1Schematic diagram of gas flow in flow tube |
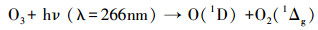 | (2) |
 | (3) |
 | (4) |
图 2(Fig. 2)
 |
| 图 2 OH自由基总反应性测定装置示意图 Fig. 2Schematic diagram of laser-induced fluorescence OH reactivity instrument |
本装置的气路系统以流动管作为主体, 其流量根据实验情况而定, 一般为15~25 L·min-1, 管内为常压.环境大气采入流动管后经约50 cm到达检测腔上方的锥形采样嘴处, 通过锥形采样嘴进入检测腔的气体样品流量为3 L·min-1.荧光检测腔内的压强则通过一台真空泵控制, 保持在3.5 mbar.荧光腔和遮光臂的两端分别注入固定流量的高纯氮气以防止样品气体污染荧光检测腔.
由于O3是总反应性测定装置的OH自由基源, 当外界O3浓度低于反应所需浓度时, 需要对其进行人工补充.因此, 在采样管路的末端处使用一台臭氧测定仪实时监测流动管内臭氧浓度, 当采样空气中O3的浓度低于10 ppbv时便进行补充, 保证流动管中OH自由基的生成浓度数量级保持在108~109 molecule·cm-3之间.经实验室测试, 该系统对kOH的测量灵敏度为1.2 s-1, 时间分辨率为5 min.
2.3 痕量物质的测量方法本次观测中其他痕量物质组分测量所使用的仪器及相关技术参数如表 1所示.其中, 常规气体的测量分别采用49i臭氧(O3)分析仪、42i一氧化氮(NO)-二氧化氮(NO2)-氮氧化物(NOx)分析仪和48i一氧化碳测定仪(CO)(Thermo Scientific, 美国).VOCs作为OH自由基总反应性的主要组成部分, 则是通过气相色谱质谱测定, 仪器的预浓缩系统由武汉市天虹仪表有限责任公司制造, GCMS-5977E分析系统由美国安捷伦公司生产, 在进样解析过程中, 目标化合物一路通过色谱柱进入到GC-FID检测器检出, 主要分析C2~C5的低碳VOCs;另一路则用于分析C5 ~ C12的高碳VOCs, 进入到MS检测器被检出.
表 1(Table 1)
| 表 1 观测站点使用仪器及技术参数 Table 1 Technical parameters of instruments used in campaign | ||||||||||||||||||||||||||||
表 1 观测站点使用仪器及技术参数 Table 1 Technical parameters of instruments used in campaign
| ||||||||||||||||||||||||||||
3 结果与讨论(Results and discussion)3.1 测量结果于2018年秋季采用OH自由基总反应性测定装置对深圳进行观测, 旨在进一步深入研究整个深圳地区的臭氧反应机制, 同时对臭氧污染较为突出的珠三角地区提出针对性治理建议.图 3是观测期间主要的气象参数与无机气体的时间序列, 测量时间为10月1—18日, 期间温度变化范围为18~32 ℃, 高温天气主要出现在观测前期时段, 相对湿度变化范围为25%~99%, 风向以东北风和南风为主.一次污染物中, CO浓度变化幅度较小, 基本维持在0.3~0.6 ppmv之间, 但在10月16日16:00 CO浓度小时均值达到观测期间的最高值0.71 ppmv(1.3 mg·m-3), 未超过国家一级标准(10 mg·m-3), 随后因为降水影响浓度迅速下降.NO与NO2的浓度水平则呈现出小范围的波动, NO2的浓度高值基本保持在40 ppbv以下, NO只在10月10日这一天的7:00—9:00间出现了高达80 ppbv以上的峰值, 此种情况可能是受站点周边机动车源的影响, 此时O3的浓度也较低, 不能被消耗掉的NO迅速累积达到高值.二次污染物中O3的浓度变化范围较大, 日间最高值可达到120 ppbv, 但观测后期由于多次冷锋过境的影响, 光化学反应缓慢, 臭氧浓度呈现出整体走低的趋势, 全天浓度基本保持在40 ppbv以下.
图 3(Fig. 3)
 |
| 图 3 气象与无机气体参数时间序列 Fig. 3Time series of meteorological parameters and inorganic gases |
图 4为观测期间主要活性VOCs浓度及总VOCs浓度时间序列图, 其中, 10月6—9日、16日的VOCs数据缺失主要是由于GC-MS仪器发生故障所致, 未能正常测量.总VOCs浓度水平维持在10~60 ppbv之间, 在10月10日及16日期间分别经历了两次浓度达到60 ppbv左右的高值后又逐渐走低的过程.作为天然源的异戊二烯在观测前期表现出较为明显的日变化规律, 午间峰值可达到3 ppbv, 后期由于光照、气温等气象条件的影响, 浓度水平维持在0.5 ppbv以下.甲醛由于有着显著的二次生成贡献, 基本表现出日间较高、夜间较低的日变化特征, 观测后期同样受到气象条件影响, 整体浓度水平较之前有所下降.烯烃类物质的浓度波动较大, 但在16日后期苯乙烯和丙烯的浓度较之前有明显的升高, 峰值均可达到1 ppbv以上.
图 4(Fig. 4)
 |
| 图 4 主要活性VOCs浓度及总VOCs浓度时间序列 Fig. 4Time series of important VOCs contributing to the OH reactivity and total VOCs |
观测期间OH自由基总反应性水平在10~30 s-1之间波动, 在以往的观测中总反应性高值一般出现在夜间(Lu et al., 2013; Fuchs et al., 2017), 但本次观测中kOH的高值却出现在白天, 这主要是受到日间天然源强烈排放的影响.结合国内外城市与郊区站点的多次观测结果, 全球OH自由基总反应性水平测量结果如表 2所示.与其他站点的结果进行对比后可以看出, 此次观测的总反应性处于一个相对较低的水平, 其测量值更加接近一些郊区站点的kOH值.
表 2(Table 2)
| 表 2 全球城市及郊区站点OH自由基总反应性测量结果 Table 2 List of OH reactivities measured in urban and suburban areas | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 2 全球城市及郊区站点OH自由基总反应性测量结果 Table 2 List of OH reactivities measured in urban and suburban areas
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
OH自由基总反应性日变化特征如图 5所示, 可以看出其日变化规律并不显著, 基本保持在16 s-1左右, 只有早高峰和夜间出现了两个较小的峰值, 与NO2的日变化走势大致相同.这与2014年夏季广东鹤山观测中表现出的总反应性日变化特征较为一致, 其总反应性日平均值为20 s-1左右, 但该站点污染时段总反应性水平较高, 峰值可达60 s-1以上;2006年广东后花园站点在整个观测期间OH自由基总反应性的变化范围为10~120 s-1, 同时表现出较为明显的日变化特征, 其夜间总反应性水平较高, 在早6:00达到一个峰值, 随后持续下降.而造成这几个站点观测结果有所差异的主要原因是选点类型不同, 其影响气团也不同, 虽然3个站点均为郊区站点, 远离典型工业园区和主要交通道路, 但相比之下后花园和鹤山站点受到人为排放的影响更大, 在生物排放的共同作用下, 其整体总反应性水平较高, 且表现出了更为明显的日变化特征.
图 5(Fig. 5)
 |
| 图 5 OH自由基总反应性日变化特征 Fig. 5Diurnal variation of total OH reactivities |
对常规气体参数分时段计算日变化特征, 结果如图 6所示.综合气象条件与常规污染物特征, 可将观测数据可大致分为3个时段:EP1(臭氧污染时段1)、EP2(臭氧污染时段2)、EP3(非臭氧污染时段).EP1期间主要受到台风“康妮”外围环流的影响, 日间光照与扩散条件良好, 夜间主要以静小风为主.O3、NOx与CO表现出明显的日变化特征, O3在每日下午达到日最大值(均值82.9 ppbv), 其中, 10月4日的O3日最大8 h均值达170.4 μg·m-3, 高于国家二级标准(160 μg·m-3), 其余时间也均超过国家一级标准(100 μg·m-3);在早高峰时段CO和NO都达到日最大值(均值0.47 ppmv与4.9 ppbv).EP1阶段光化学过程强烈, 温度较高, 日均PM2.5与由天然源排放的异戊二烯浓度也显著高于另外两个时段.EP2和EP3期间分别受两次冷锋过境的影响.10月10日有短时局地强降雨, 冷锋过境后温度迅速回升, 相对湿度总体升高, 日变化幅度减弱, 基本维持在80%以上.EP2期间O3在14:00左右达到日最大值(均值49.6 ppbv), 该时段内10月12日和13日的O3日最大8 h均值分别为101.6 μg·m-3和102.8 μg·m-3, 高于国家一级标准, 其余时间则略低于一级标准, 因此, EP2仍处于臭氧污染时期, 但污染水平较EP1期间有所下降;CO和NO的日变化特征不够明显, CO的日最大值未出现在早高峰时段, 而是出现在夜间23:00(均值0.43 ppmv), 同时NO早高峰出现的时间与CO相比也有所不同, 滞后了近2 h.EP3阶段中经历了新一轮的冷锋过境, 3 d内都有不同程度的降水过程, 日均相对湿度均在80%以上, 温度较EP1和EP2有明显降低.该阶段日照不强, 光化学过程较弱, O3的最大小时均值仅为27.6 ppbv.NO的日最大值出现在上午10:00前后, 与EP1早高峰时段明显不同, 可判断为受到气团传输影响.CO在冷锋过境前浓度迅速升高, 16日16:00达到观测期间的最高值0.71 ppmv, 同样表明受到了外来气团传输的影响.此阶段一次污染物浓度显著高于EP1与EP2, 二次氧化过程影响较小.通过上述分析可以看到3个时段的气象条件与常规污染物特征都有着较为明显的差别, 因此, 将观测期间的相关数据按3个时段进行划分, 从而对OH自由基总反应性进行进一步的讨论.
图 6(Fig. 6)
 |
| 图 6 不同时段O3-NO-NO2-CO及OH自由基总反应性日变化特征 (a.10月1—4日(EP1), b.10月11—15日(EP2), c.10月16—18日(EP3)) Fig. 6Diurnal variations of O3-NO-NO2-CO and total OH reactivities of EP1(a), EP2(b) and EP3(c) |
3.2 kOH物种组成将各物种对应的反应活性平均值与实测总反应性平均值相除, 即可得到OH自由基总反应性的物种组成.图 6的右侧部分展示了3个时段总反应性的日变化特征, 图 7则是整个观测期间及3个时段分别对应的物种组成, 结合两幅图可以看出, 整个观测期间无机物和一次排放的VOCs对总反应性的贡献较大, 特别是无机物(CO和NOx)的贡献在不同时段都占到了30%~40%左右, 这与大部分郊区站点的观测结果一致, 无机物为总反应性的主要贡献者, 在以往的观测中早高峰时段无机物的活性占比甚至可以达到60%(Ren et al., 2005).
图 7(Fig. 7)
 |
| 图 7 不同时段OH自由基总反应性物种组成 (a.EP1, b.EP2, c.EP3, d.整个观测期间) Fig. 7Composition of total OH reactivities of EP1(a), EP2 (b), EP3 (c) and the whole observation(d) |
在无机物作为主要贡献者的基础上, 一次排放的VOCs在总反应性中也有着较高的占比, 约为20%~30%.EP1期间作为天然源的异戊二烯占比突出, 达到了10%左右, 这是由于异戊二烯主要由白天植物光合作用释放产生, 而这期间整体气温偏高, 午间最高值达30 ℃以上, 且日照充足, 由此产生的异戊二烯浓度较高并在午间达到峰值, 因此从日变化特征来看, 其反应活性在午间的占比也尤为突出.这一贡献值与Elshorbany等(2012)的观测结果较为接近, 大约在6%~16%之间, 但与2006年广东后花园(Lou et al., 2010)的观测结果相比, 由于后花园点位离农田和森林较近, 异戊二烯贡献了峰值近12 s-1的反应活性, 其日间的占比可以达到70%, 因此, 异戊二烯在总反应性中的占比大小也会受到点位的影响.EP3期间芳香烃的贡献则非常显著, 占到20%, 与珠三角地区相比较为一致, 其秋季VOCs组分中活性较强的物质一般是芳香烃与烯烃(邓雪娇等, 2010);与全国其他地区相比, 夏、秋季节除京津冀地区主要以烯烃贡献为主外, 长三角地区和成渝地区也均有着较高的芳香烃水平(蒋美青等, 2018), 这与两种物质的OH反应速率常数较高有着一定的关系, 这种情况下单位浓度芳香烃与烯烃对总反应性的贡献远高于其他物质(Wang et al., 2014).
除无机物与一次排放的VOCs的主要贡献外, 以醛酮类为主的二次有机氧化物种OVOCs也值得关注, 它主要来自于长距离输送的老化气团.该物种也展现出了与异戊二烯类似的日变化规律, 即OVOCs在日间对总反应性的贡献高于夜间, 这一特点在日间二次氧化反应较为剧烈的EP1和EP2期间较为突出, 甲醛和OVOCs的平均占比达到了15%左右.就甲醛和乙醛而言, 此次观测中两种物质不仅反应活性水平高, 其浓度较其他站点也整体偏高, 这也与之前在珠三角进行的观测结果相符(Yuan et al., 2012), 而其前体物NMHCs的氧化是OVOCs的重要来源之一, 因此也在一定程度上表明观测点位处NMHCs的强烈排放(Wang et al., 2015).进一步将该占比与之前各郊区站点的观测结果进行比较可以看出, OVOCs在总反应性中的贡献也是不可忽视的.在2014年河北望都的观测中, OVOCs对总反应性的贡献甚至超过了50%, 主要以甲醛和乙醛为主(20%~25%)(Fuchs et al., 2017), 在广东鹤山观测期间其贡献也较为突出(Yang et al., 2017).而在更早的观测中, 虽然没有直接对OVOCs进行测量, 但通过模型模拟对OVOCs进行补充之后, 缺失的总反应性明显减少, 特别是后花园站点其日间活性缺失经判断全部来自于OVOCs(Lou et al., 2010), 北京榆垡站点经OVOCs约束过的模拟值同样可以很好地吻合实测活性值(Lu et al., 2013).而参与模型模拟的物质通常有甲醛、乙醛、乙二醛、MVK及MACR等, 它们通常也是受人为源影响较大的观测地区的主要OVOCs物种, 此类观测地区包括北京(Shao et al., 2009)、东京(Yoshino et al., 2012)及伦敦(Whalley et al., 2016)等地, 这同时也证明了这些羰基化合物是污染边界层中与OH自由基反应的主要物质.
整个观测期间OH自由基总反应性的测量平均值为16.8 s-1, 而通过CO、NOx及碳氢化合物等实测活性物质浓度汇总计算所得的总反应性值为12.9 s-1, 由此计算得到整个观测期间的缺失活性占比约为23%.但根据图 7分时段进行计算后发现, 不同时段的活性缺失程度依然存在着一定差异.在EP1和EP2期间由于光照条件良好, 日间异戊二烯浓度较高且其反应速率常数较大, 使得日间总反应性的计算值基本接近于实测值, 因此主要的缺失来自于夜间, 其全天活性缺失占比均接近于30%;而EP3期间全天的活性缺失平均不超过16%, 此时芳香烃的本地排放较为强烈, 总反应性水平较之前有明显提高.
3.3 活性缺失的相关性分析根据3个观测时段的总反应性日变化来看, 其缺失现象主要发生在夜间及早晚高峰时段, 此时二次产物的浓度往往较低, 相应的一次排放则较强, 同时根据观测期间OC/EC的比值计算二次生成的SOC对于OC的贡献, 其公式(郭安可等, 2017)为:
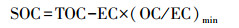 | (5) |
由于不同排放源的VOCs化学组成存在差异, 在对观测期间的活性缺失进行相关性分析前, 需要先通过VOCs物种的比值对3个时段大气中VOCs的来源进行初步定性分析.文献中一般采用甲苯与苯的比值(T/B)来进行判断(Mo et al., 2017), 其机动车排放与典型城市的特征值一般在2~3之间(Tang et al., 2007), 工业区环境大气中测到的比值为6.0~6.9(Chan et al., 2006), 溶剂涂料行业中的T/B值为11.5(Yuan et al., 2010).计算结果如图 8所示, 3个时段T/B的值分别为5.41、6.70、14.61, 与机动车排放特征值相比明显偏高, 这是由于甲苯除了来自机动车排放外, 溶剂涂料的使用是其更为主要的一类来源, T/B值越高则说明受到溶剂涂料的影响越大(Chan et al., 2006).EP3与EP1、EP2相比特征值显著不同, 更接近涂料溶剂行业的比值, 因此, 该时段内溶剂涂料的排放有着更为明显的影响.其次, 由于这3个时段甲苯与苯的相关性一般, T/B的波动也较大, 说明VOCs的各排放源占比产生了一定的变化.虽然总体排放结构在短时期内不会发生改变, 但考虑到3个时段内不同气象因素对于传输的影响, 其排放特征还是存在一定差异.
图 8(Fig. 8)
 |
| 图 8 不同时段甲苯与苯的相关关系 (a.EP1, b.EP2, c.EP3) Fig. 8Correlation between toluene and benzene of EP1(a), EP2 (b) and EP3(c) |
将VOCs一次物种的反应活性日变化值按照缺失现象主要发生的夜间和早晚高峰时段(0:00—9:00及18:00—24:00)提出, 在EP1、EP2与EP3三个时段内, 分别将其与对应时段的缺失活性值进行相关性分析, 结果如图 9所示, 相关性较好则表明该物种所示踪的排放源对于缺失活性有比较重要的贡献.3个时段最终筛选出的物种为苯乙烯、丙烯和戊烯, 对应的相关系数分别为0.92、0.86和0.81.文献中苯乙烯和丙烯常分别作为溶剂涂料(陆思华等, 2006)和石化工业的示踪物, 而戊烯则一般为机动车排放源(周炎等, 2017)的示踪物, 考虑到机动车排放源一般有LPG车辆、汽油车和柴油车这3类, 而丙烷和乙烷是LPG和NG的重要成分, 可通过其比值P/E将机动车排放源进一步细分.研究发现, LPG车辆尾气中P/E的值约为3(王鸣等, 2018), 而整个观测期间P/E约为2, 在机动车排放与活性缺失相关性较好的时段内, 该比值可以达到3.2左右.由此可以判断机动车排放源中, 活性缺失主要来自于LPG机动车排放的一次污染物及相关二次产物.综上所述, 由于3类烯烃的排放源实际上并不唯一, 在上述3类排放源中3种物质均占有一定的比重, 因此, 我们认为溶剂涂料、石化工业及LPG机动车排放这3类源是整个观测期间活性缺失的主要贡献者.同时在此前的观测中, Kovacs等(2003)等在美国纳什维尔观测中通过隧道实验发现缺失活性来源于短寿命VOCs;2004年春季在英国的观测中, Lee(2009)曾发现39%左右的活性缺失, 通过模型模拟发现缺失大部分来源于高分子芳香类化合物;但更多的观测结果表明缺失活性很大程度上可能来自于二次氧化产物, 此次观测中缺失活性却与二次产物的相关性较差, 对于这部分来源, 还需要模型模拟来进行进一步的探究.
图 9(Fig. 9)
 |
| 图 9 不同时段反应缺失活性与相关来源物种活性相关关系 (a.EP1, b.EP2, c.EP3) Fig. 9Correlation between missing reactivities and reactivities calculated from source species of EP1(a), EP2 (b) and EP3(c) |
4 结论(Conclusions)1) 观测期间, 深圳的OH自由基总反应性水平整体较低, 在10~30 s-1之间波动, 日变化特征不明显, 其中, 无机物对于总反应性有着最主要的贡献, 3个时段内CO和NOx在整体中的占比均接近40%左右.
2) 一次排放的VOCs对于总反应性的贡献仅次于无机物, 占到20%~30%;在光照条件较好的日间, 由天然源排放的异戊二烯有着较为明显的占比, 本地排放较为强烈的时段则主要以芳香烃的贡献为主, 同时在二次氧化反应较为剧烈的时段内OVOCs的贡献也不容忽视.
3) 整个观测期间OH自由基总反应性的缺失为23%左右, 主要缺失时段为夜间和早晚高峰, 缺失活性主要来源于溶剂涂料、石化工业及LPG机动车排放.
参考文献
| Calpini B, Jeanneret F, Bourqui M, et al. 1999. Direct measurement of the total reaction rate of OH in the atmosphere[J]. Analusis, 27(4): 328–336.DOI:10.1051/analusis:1999270328 |
| Chan L, Chu K, Zou S, et al. 2006. Characteristics of nonmethane hydrocarbons (NMHCs) in industrial, industrial-urban, and industrial-suburban atmospheres of the Pearl River Delta (PRD) region of south China[J]. Journal of Geophysical Research - Atmospheres, 111(D11): D11304.DOI:10.1029/2005JD006481 |
| 邓雪娇, 王新明, 赵春生, 等. 2010. 珠江三角洲典型过程VOCs的平均浓度与化学反应活性[J]. 中国环境科学, 2010, 30(9): 1153–1161. |
| Dolgorouky C, Gros V, Sarda-Esteve R, et al. 2012. Total OH reactivity measurements in Paris during the 2010 MEGAPOLI winter campaign[J]. Atmospheric Chemistry and Physics, 12(20): 9593–9612.DOI:10.5194/acp-12-9593-2012 |
| Ehhalt D H. 1999. Photooxidation of trace gases in the troposphere[J]. Physical Chemical Physics, 1(24): 5401–5408.DOI:10.1039/a905097c |
| Elshorbany Y F, Kleffmann J, Hofzumahaus A, et al. 2012. HOx budgets during HOxComp:A case study of HOx chemistry under NOx-limited conditions[J]. Journal of Geophysical of Geophysical Research-Atmospheres, 117(D3).DOI:10.1029/2011JD017008 |
| Fuchs H, Tan Z, Lu K, et al. 2017. OH reactivity at a rural site (Wangdu) in the North China Plain:contributions from OH reactants and experimental OH budget[J]. Atmospheric Chemistry and Physics, 17(1): 645–661.DOI:10.5194/acp-17-645-2017 |
| Goldstein A H, Galbally I E. 2007. Known and unexplored organic constituents in the earth′s atmosphere[J]. Environmental Science & Technology, 41(5): 1514–1521. |
| 郭安可, 郭照冰, 张海潇, 等. 2017. 南京北郊冬季PM2.5中水溶性离子以及碳质组分特征分析[J]. 环境化学, 2017, 36(2): 248–256. |
| 蒋美青, 陆克定, 苏榕, 等. 2018. 我国典型城市群O3污染成因和关键VOCs活性解析[J]. 科学通报, 2018, 63(12): 1130–1141. |
| Kovacs T A, Brune W H. 2001. Total OH Loss Rate Measurement[J]. Journal of Atmospheric Chemistry, 39(2): 105–122.DOI:10.1023/A:1010614113786 |
| Kovacs T A, Brune W H, Harder H, et al. 2003. Direct measurements of urban OH reactivity during Nashville SOS in summer 1999[J]. Journal of Environmental Monitoring, 5(1): 68–74.DOI:10.1039/b204339d |
| Lee J D, Young J C, Read K A, et al. 2009. Measurement and calculation of OH reactivity at a United Kingdom coastal site[J]. Journal of Atmospheric Chemistry, 64(1): 53–76.DOI:10.1007/s10874-010-9171-0 |
| Liu Y, Shao M, Lu S, et al. 2008. Volatile organic compound (VOC) measurements in the pearl river delta (PRD) region, China[J]. Atmospheric Chemistry and Physics, 8(6): 1531–1545.DOI:10.5194/acp-8-1531-2008 |
| Lou S, Holland F, Rohrer F, et al. 2010. Atmospheric OH reactivities in the Pearl River Delta - China in summer 2006:measurement and model results[J]. Atmospheric Chemistry and Physics, 10(22): 11243–11260.DOI:10.5194/acp-10-11243-2010 |
| Lu K D, Hofzumahaus A, Holland F, et al. 2013. Missing OH source in a suburban environment near Beijing:observed and modelled OH and HO2 concentrations in summer 2006[J]. Atmospheric Chemistry and Physics, 13(2): 1057–1080.DOI:10.5194/acp-13-1057-2013 |
| 陆思华, 白郁华, 张广山, 等. 2006. 大气中挥发性有机化合物(VOCs)的人为来源研究[J]. 环境科学学报, 2006, 26(5): 757–763.DOI:10.3321/j.issn:0253-2468.2006.05.010 |
| Mo Z, Shao M, Lu S, et al. 2017. Characterization of non-methane hydrocarbons and their sources in an industrialized coastal city, Yangtze River Delta, China[J]. Science of the Total Environment, 593-594: 641–653.DOI:10.1016/j.scitotenv.2017.03.123 |
| Muller J B A, Elste T, Plass-Dülmer C, et al. 2018. A novel semi-direct method to measure OH reactivity by chemical ionization mass spectrometry (CIMS)[J]. Atmospheric Measurement Techniques, 11(7): 4413–4433.DOI:10.5194/amt-11-4413-2018 |
| Ren X, Brune W H, Cantrell C A, et al. 2005. Hydroxyl and peroxy radical chemistry in a rural area of central pennsylvania:Observations and model comparisons[J]. Journal of Atmospheric Chemistry, 52(3): 231–257.DOI:10.1007/s10874-005-3651-7 |
| Shao M, Lu S, Liu Y, et al. 2009. Volatile organic compounds measured in summer in Beijing and their role in ground‐level ozone formation[J]. Journal of Geophysical Research, 114: 6–19. |
| Sinha V, Williams J, Crowley J N, et al. 2008. The comparative reactivity method - a new tool to measure total OH reactivity in ambient air[J]. Atmospheric Chemistry and Physics, 8(8): 2213–2227.DOI:10.5194/acp-8-2213-2008 |
| Stone D, Whalley L K, Heard D E. 2012. Tropospheric OH and HO2 radicals:field measurements and model comparisons[J]. Chem Soc Rev, 41(19): 6348–6404.DOI:10.1039/c2cs35140d |
| Stone D, Whalley L K, Ingham T, et al. 2016. Measurement of OH reactivity by laser flash photolysis coupled withlaser-induced fluorescence spectroscopy[J]. Atmospheric Measurement Techniques, 9(7): 2827–2844.DOI:10.5194/amt-9-2827-2016 |
| Tang J H, Chan L Y, Chan C Y, et al. 2007. Characteristics and diurnal variations of NMHCs at urban, suburban, and rural sites in the Pearl River Delta and a remote site in South China[J]. Atmospheric Environment, 41(38): 8620–8632.DOI:10.1016/j.atmosenv.2007.07.029 |
| Wang M, Chen W, Shao M, et al. 2015. Investigation of carbonyl compound sources at a rural site in the Yangtze River Delta region of China[J]. Journal of Environmental Sciences, 28: 128–136.DOI:10.1016/j.jes.2014.12.001 |
| Wang M, Shao M, Chen W, et al. 2014. A temporally and spatially resolved validation of emission inventories by measurements of ambient volatile organic compounds in Beijing, China[J]. Atmospheric Chemistry and Physics, 14(12): 5871–5891.DOI:10.5194/acp-14-5871-2014 |
| Whalley L K, Stone D, Bandy B, et al. 2016. Atmospheric OH reactivity in central London:observations, model predictions and estimates of in situ ozone production[J]. Atmospheric Chemistry and Physics, 16(4): 2109–2122.DOI:10.5194/acp-16-2109-2016 |
| 王鸣, 陈文泰, 陆思华, 等. 2018. 我国典型城市环境大气挥发性有机物特征比值[J]. 环境科学, 2018, 39(10): 4393–4399. |
| Yang Y, Shao M, Ke?el S, et al. 2017. How the OH reactivity affects the ozone production efficiency:case studies in Beijing and Heshan, China[J]. Atmospheric Chemistry and Physics, 17(11): 7127–7142.DOI:10.5194/acp-17-7127-2017 |
| Yoshino A, Nakashima Y, Miyazaki K, et al. 2012. Air quality diagnosis from comprehensive observations of total OH reactivity and reactive trace species in urban central Tokyo[J]. Atmospheric Environment, 49: 51–59.DOI:10.1016/j.atmosenv.2011.12.029 |
| Yuan B, Chen W, Shao M, et al. 2012. Measurements of ambient hydrocarbons and carbonyls in the Pearl River Delta (PRD), China[J]. Atmospheric Research, 116: 93–104.DOI:10.1016/j.atmosres.2012.03.006 |
| Yuan B, Shao M, Lu S, et al. 2010. Source profiles of volatile organic compounds associated with solvent use in Beijing, China[J]. Atmospheric Environment, 44(15): 1919–1926.DOI:10.1016/j.atmosenv.2010.02.014 |
| 周炎, 岳玎利, 张涛. 2017. 春季广州城区空气中VOCs来源解析[J]. 环境监控与预警, 2017, 9(1): 42–47.DOI:10.3969/j.issn.1674-6732.2017.01.014 |
