摘要/Abstract
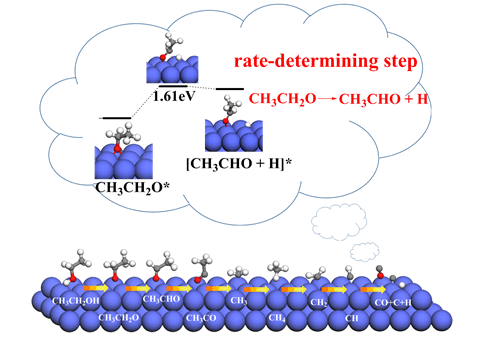
采用密度泛函理论和周期平板模型相结合的方法针对Co(111)表面上乙醇脱氢反应的反应机理进行了细致的研究,同时,对反应过程中涉及到的各个物种在表面上不同吸附位(顶位(top),桥位(bridge),三重空穴位(fcc和hcp))的吸附模型进行了结构优化以及相关能量的计算,确定了各物种的最佳吸附位点.研究结果表明,CH3CH2OH在Co(111)表面的脱氢反应可具体描述为三条反应路径:反应路径I为CH3CH2OH逐步脱氢经由中间体CH3CHO,最终生成CH4和CO的反应;反应路径Ⅱ为CH3CH2OH脱氢产生的CH3CH2O基和CH3CHO相互作用通过CH3COOH分子最终生成CH4和CO2的反应;反应路径Ⅲ为CH3CH2O基和CH3CO基作用生成CH3COOC2H5的过程,其中,反应路径I为最优路径(CH3CH2OH→CH3CH2O→CH3CHO→CH3CO→CH3+CO→CH2→CH→CH4+CO+C+H),该反应路径中的CH3CH2O基脱氢生成CH3CHO为速控步骤,反应能垒为1.61 eV.
关键词: 第一性原理, 乙醇, Co(111)表面, 脱氢, 吸附
The detailed reaction mechanism of ethanol dehydrogenation on Co(111) surface was studied using the density functional theory (DFT) and slab periodic model. The structures and energies of the species involved in the reaction adsorbed on different adsorption sites (top, fcc, hcp and bridge sites) of the surface were calculated and compared. The calculated results show that ethanol adsorbs weakly on the Co(111) surface. CH3CH2O, CH and C prefer hcp sites with adsorption energies of -2.72, -6.85, and -6.92 eV, respectively. CH3CHO adsorbs weakly at the bridge-η1(O)-η1(Cα) site with adsorption energy of -0.47 eV. CH3CO and CH2 adsorb stably on Co(111) surface through their unsaturated C atoms with binding energies of -2.31 and -3.90 eV, respectively. CH3 and CH4 prefer to locate at top sites through the C atom with adsorption energies of -1.95 and -0.12 eV, respectively. CO and H are bind stably at fcc sites with binding energies of -1.62 and -2.77 eV, respectively. Due to the complexity of the decomposition of ethanol, the scissions of O-H, C-H, C-O and C-C bonds of CH3CH2OH were examined. The results show that ethanol decomposition on Co(111) surface starts with the scission of the O-H bond, and the dehydrogenation reaction of ethanol on Co(111) surface can be described as three reaction pathways:Path I is the gradual dehydrogenation of CH3CH2OH via intermediate CH3CHO, which ultimately produces CH4 and CO; Path Ⅱ is the reaction of CH3CH2O and CH3CHO which were generated by dehydrogenation of ethanol, to form CH4 and CO2 via CH3COOH intermediate; Path Ⅲ is the process of CH3CH2O reacts with CH3CO to generate CH3COOC2H5. On the basis of our computational results, Path I (CH3CH2OH→CH3CH2O→CH3CHO→CH3CO→CH3+CO→CH2→CH→CH4+CO+C+H) is more favorable than Paths Ⅱ and Ⅲ and the dehydrogenation of CH3CH2O to CH3CHO is the rate-determining step with a reaction energy barrier of 1.61 eV.
Key words: first principle, ethanol, Co(111) surface, dehydrogenation, adsorption
PDF全文下载地址:
点我下载PDF
