药物和靶标之间存在错综复杂的网络关系。随着网络药理学等概念的快速发展,药物发现模式开始从“一个药物→一个靶标 → 一种疾病”的线性模式,逐渐转向“多个药物 → 多个靶标 → 多种疾病”的网络模式。通过基于网络的计算方法,可有效预测药物和类药化合物预测潜在的靶标,或为感兴趣的靶标预测潜在的活性化合物。然而,现有基于网络的药物—靶标相互作用预测方法只能预测相互作用的有无,尚未考虑相互作用的强弱,难以有效预测对特定靶标具有高活性的化合物。
为了克服这一局限性,本研究提出了一种名为加权的基于子结构-药物-靶标网络推理(wSDTNBI)的新方法。作为一种基于网络的方法,wSDTNBI方法不仅保持了此类方法已有优势,既不依赖于靶标三维结构,也不依赖于阴性样本。该方法还合理引入了能够反映药物-靶标相互作用强弱的结合亲和力数据,通过一种将网络推理和相似性推理相结合的“双管齐下”途径,使得预测打分可以反映相互作用强弱,从而快速、高效地发现高活性的化合物。在本研究中,wSDTNBI方法仅耗费数十秒时间,即完成了超过一万三千个化合物和近两千个人类靶标之间的计算预测,即可同时针对这些靶标开展了虚拟筛选。通过对部分预测结果进行多种类型的体外实验,本研究成功发现了7个新的维甲酸相关孤儿受体γt(RORγt)反向激动剂。其中,乌索酮酸和齐墩果酮酸两个天然产物的活性尤为突出,对预测靶标RORγt 的半数抑制浓度(IC50)分别达到了10 nM和0.28 μM。本研究通过等温量热滴定实验获得了活性化合物与靶标蛋白的结合常数,通过复合物晶体结构解析了活性化合物与靶标蛋白的相互作用结构特征。此外,乌索酮酸和齐墩果酮酸在动物实验中也表现出了良好的治疗效果,有望成为靶向RORγt治疗多发性硬化症等自身免疫疾病的先导化合物。
相比于近年来针对同一靶标的基于结构或基于深度学习的虚拟筛选研究,本研究不仅具有更高的成功率,也有助于获得更高活性的潜在药物分子。目前,wSDTNBI方法已经整合到唐赟教授课题组的在线服务系统NetInfer(网址:http://lmmd.ecust.edu.cn/netinfer)中,供国内外用户免费使用,可以为针对不同疾病的多类型靶标的药物发现研究提供帮助。
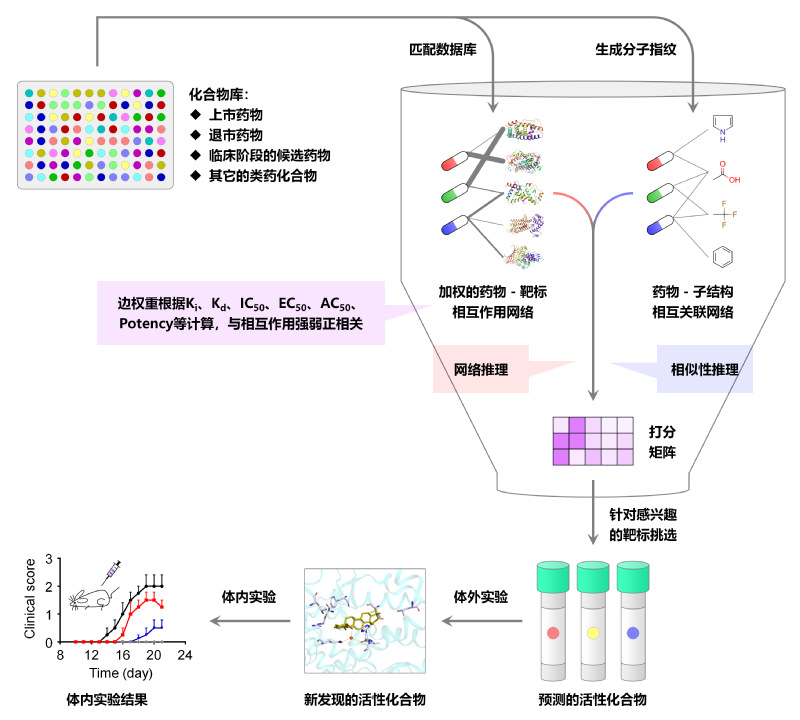
?图片说明:使用wSDTNBI方法进行活性化合物筛选的示意图
论文的第一作者是我校药学院的吴曾睿博士和研究生马辉,通讯作者为唐赟教授和黄瑾教授。该项研究得到了国家重点研发计划、国家自然科学基金、上海市细胞代谢光遗传学技术前沿科学研究基地、上海市“超级博士后”激励计划、上海市青年科技英才扬帆计划、中国博士后科学基金的经费支持。
原文链接:https://pubs.rsc.org/en/content/articlelanding/2022/sc/d1sc05613a
