罗静初
(北京大学 生命科学学院,北京大学生物信息中心,北京 100871)
摘要:
本文介绍欧洲分子生物学开放软件包EMBOSS序列分析程序应用实例。第1节简单介绍EMBOSS软件包的概况和基本用法。第2节介绍格式转换、序列提取、序列变换和序列显示等常用序列处理程序。第3节介绍序列比对程序,包括双序列比对、多序列比对和点阵图程序。第4节介绍常用核酸序列分析程序,可用于核苷酸组分统计、开放读码框分析、CpG岛识别、密码子使用统计和重复序列寻找等。第5节介绍常用蛋白质序列分析程序,包括氨基酸组分统计、序列特征位点识别、二级结构分析等。文中结合教学实例,选择部分常用程序,给出具体运行方式,并扼要说明分析结果的生物学意义。文末对程序运行过程中需要注意的地方加以讨论,并用表格列出部分常用程序的名称和用途,以便读者查阅。
关键词: EMBOSS软件包 双序列比对 多序列比对 点阵图 核酸序列分析 蛋白质序列分析
DOI:10.12113/202008002
分类号:Q349+.53
文献标识码:A
基金项目:
Application examples of EMBOSS sequence analysis program
LUO Jingchu
(College of Life Sciences and Center for Bioinformatics, Peking University, Beijing 100871, China)
Abstract:
The aim of this paper is to introduce the European Molecular Biology Open Software Suite (EMBOSS) with practical application examples. In the first section, a brief overview about EMBOSS is given and general usages of the programs are described. The second section introduces tools for sequence format conversion, sequence retrieval, manipulation, and display. The third section presents sequence alignment programs including pairwise and multiple sequence alignment as well as dot-plot. The fourth section reviews commonly used nucleotide sequence analyses, which can be used in composition statistics, open reading frame analysis, CpG island prediction, codon usage statistics, and repeat sequence identification. Protein sequence analysis programs such as amino acid composition calculation, sequence motif discovery, and secondary structure analysis are summarized in the last section. Application examples for some commonly used programs are described based on teaching experiences. The specific operation steps to run the programs and the biological significance of the analysis results are elucidated.Lastly, special notes are discussed for the purpose of better use of the programs, and a summary table containing the names and usages of some programs is given.
Key words: EMBOSS Pairwise sequence alignment Multiple sequence alignment Dot-plot Nucleotide sequence analysis Protein sequence analysis
罗静初. EMBOSS软件包序列分析程序应用实例[J]. 生物信息学, 2021, 19(1): 1-25. DOI: 10.12113/202008002.

LUO Jingchu. Application examples of EMBOSS sequence analysis program[J]. Chinese Journal of Bioinformatics, 2021, 19(1): 1-25. DOI: 10.12113/202008002.
作者简介 罗静初,男,教授,研究方向:实用生物信息技术. E-mail: luojc@pku.edu.cn 文章历史 收稿日期: 2020-08-04 修回日期: 2020-08-18
Contents Abstract Full text Figures/Tables PDF
EMBOSS软件包序列分析程序应用实例
罗静初

北京大学 生命科学学院,北京大学生物信息中心, 北京 100871
收稿日期: 2020-08-04; 修回日期: 2020-08-18
作者简介:罗静初,男,教授,研究方向:实用生物信息技术. E-mail: luojc@pku.edu.cn
摘要: 本文介绍欧洲分子生物学开放软件包EMBOSS序列分析程序应用实例。第1节简单介绍EMBOSS软件包的概况和基本用法。第2节介绍格式转换、序列提取、序列变换和序列显示等常用序列处理程序。第3节介绍序列比对程序,包括双序列比对、多序列比对和点阵图程序。第4节介绍常用核酸序列分析程序,可用于核苷酸组分统计、开放读码框分析、CpG岛识别、密码子使用统计和重复序列寻找等。第5节介绍常用蛋白质序列分析程序,包括氨基酸组分统计、序列特征位点识别、二级结构分析等。文中结合教学实例,选择部分常用程序,给出具体运行方式,并扼要说明分析结果的生物学意义。文末对程序运行过程中需要注意的地方加以讨论,并用表格列出部分常用程序的名称和用途,以便读者查阅。
关键词: EMBOSS软件包 双序列比对 多序列比对 点阵图 核酸序列分析 蛋白质序列分析
Application examples of EMBOSS sequence analysis program
LUO Jingchu
College of Life Sciences and Center for Bioinformatics, Peking University, Beijing 100871, China
Abstract: The aim of this paper is to introduce the European Molecular Biology Open Software Suite (EMBOSS) with practical application examples. In the first section, a brief overview about EMBOSS is given and general usages of the programs are described. The second section introduces tools for sequence format conversion, sequence retrieval, manipulation, and display. The third section presents sequence alignment programs including pairwise and multiple sequence alignment as well as dot-plot. The fourth section reviews commonly used nucleotide sequence analyses, which can be used in composition statistics, open reading frame analysis, CpG island prediction, codon usage statistics, and repeat sequence identification. Protein sequence analysis programs such as amino acid composition calculation, sequence motif discovery, and secondary structure analysis are summarized in the last section. Application examples for some commonly used programs are described based on teaching experiences. The specific operation steps to run the programs and the biological significance of the analysis results are elucidated.Lastly, special notes are discussed for the purpose of better use of the programs, and a summary table containing the names and usages of some programs is given.
Key Words: EMBOSS Pairwise sequence alignment Multiple sequence alignment Dot-plot Nucleotide sequence analysis Protein sequence analysis
1 概述1.1 简介欧洲分子生物学开放软件包(European Molecular Biology Open Software Suite, EMBOSS)诞生于上个世纪九十年代末,是较早投入使用的大型生物信息学开放软件包,包括300多个程序,主要用于核酸和蛋白质序列分析[1-3]。EMBOSS是欧洲分子生物学网络组织(European Molecular Biology Network, EMBnet)启动的以欧洲国家为主的国际合作项目,主要发起人和开发者为Peter Rice和Alan Bleasby。EMBOSS是开源软件包,源代码完全公开,任何人均可免费获取、安装、使用和修改,并可进行二次开发,例如开发浏览器用户界面等。EMBOSS官方网站除提供软件下载外,还提供用户文档、使用教程、开发指南和常见问题解答等相关资料。
2001年起,笔者在北京大学开设“实用生物信息技术”研究生课程[4],曾介绍EMBOSS软件包中部分常用程序。2020年新冠病毒疫情期间,为北京大学生物信息学专业本科生开设“Linux基础及其在生物信息领域中的应用”线上课程,较为系统地介绍了EMBOSS软件包主要程序。
为推广EMBOSS软件包在生物信息学研究中的应用,本文基于教学中的一些实例,介绍EMBOSS软件包中主要程序的用途、用法,及其运行结果的生物学意义。文中所举实例的蛋白质序列源自国际蛋白质数据库UniProt[5],核酸序列源自美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)的核酸序列数据库GenBank和参考序列数据库RefSeq。文中对所举实例的生物学背景作了简要说明,并给出相关文献,具体细节可参阅“实用生物信息技术”课程(Applied Bioinformatics Course, ABC)教学网站(https://bigd.big.ac.cn/education/)。获中国科学院北京基因组研究所大数据中心支持,本文补充材料已上载到该所国家生物信息中心网站(https://bigd.big.ac.cn/education/)。文中所涉及的网站请参阅补充材料1,所举实例中的输入数据可通过补充材料2中的链接下载,运行结果可通过补充材料3中的链接查看。计划将文中主要程序实例改编成网络教程,并不断进行扩充和更新。
1.2 用户界面EMBOSS序列分析程序的主要使用方式有以下三种,读者可根据自己的实际情况,适当采用以下一种或几种方式。
1.2.1 命令行方式EMBOSS基于UNIX操作系统开发,所有程序均可在Linux系统上用命令行方式执行。国内外不少生物信息学相关科研机构、高等学校和公司企业的Linux服务器上装有EMBOSS软件包。读者可向服务器管理员申请Linux系统账号,登录后即可通过命令行方式运行该软件包中的程序。MacOS系统用户可下载EMBOSS源代码并编译、安装该软件包。基于Windows系统版本mEMBOSS可在磁盘操作系统(Disk Operation System, DOS)环境下以命令行方式运行。
1.2.2 图形界面命令行方式操作快速高效,是熟悉Linux系统用户的首选。不熟悉Linux系统的用户可下载基于Windows系统的mEMBOSS软件包,在装有Java运行环境的Windows系统中启动Jemboss图形界面,运行EMBOSS程序[6]。
1.2.3 浏览器界面除上述两种用户界面外,还可通过浏览器访问装有EMBOSS软件包的服务器。这类服务器提供基于EMBOSS软件包开发的用户界面,如北京大学生物信息中心开发的生物信息网上实验室WebLab,荷兰瓦格宁根大学(Wageninen University)开发的EMBOSS Explorer,以及欧洲生物信息学研究所(European Bioinformatics Institute, EBI)安装的部分EMBOSS程序(补充材料)。
1.3 运行模式上述三种用户界面中,命令行方式是最基本的运行方式,本文仅介绍这种运行方式。具体运行时,按参数选择方式的不同,可分为以下三种模式。
1.3.1 交互式第一种为交互式,运行时只输入程序名,系统给出提示符后再逐项输入需要处理的序列文件名、输出结果文件名和程序参数。参数可以有1个或多个,用户可采用系统提供的默认参数,也可自行输入。下面,我们以血红蛋白(Hemoglobin)为例,说明交互式程序运行模式。
血红蛋白是重要生物大分子,在生命科学研究历史中具有特殊地位。国际蛋白质结构数据库(Protein Data Bank, PDB)分子月报(Molecule of the Month)网站科普短文介绍了它的结构功能关系。UniProt数据库蛋白质分子精选(Protein Spotlight)网站介绍了血红蛋白研究历史。血红蛋白是第一个被确定生理功能的生物大分子,于1849年获得纯化晶体;也是第一个准确测得分子量的蛋白质、第一个在体外非细胞体系中人工合成的真核生物大分子,其编码基因的mRNA最先被分离并测序。人类基因组中有alpha和beta两大类血红蛋白编码基因,共编码9种不同的血红蛋白[4]。成人血液中的血红蛋白由两个alpha血红蛋白亚基和两个beta血红蛋白亚基组成。研究表明,小鼠alpha血红蛋白和人的alpha血红蛋白是直系同源蛋白,起源于共同祖先,其序列相似性较高。
从蛋白质数据库UniProt中下载FASTA格式的人和小鼠alpha血红蛋白氨基酸序列,分别保存为HBA_HUMAN.FASTA和HBA_MOUSE.FASTA,用EMBOSS软件包中的程序needle进行序列比对。

上述运行过程说明如下。
1) 用户输入程序名needle,按回车键后开始运行。
2) 系统给出所运行程序的简单说明Needleman-Wunsch global alignment of two sequences,并提示用户输入用于比对的序列文件名Input sequence。
3) 输入第一个序列文件名HBA_HUMAN.FASTA后系统提示输入第二个序列文件名。
4) 输入第二个序列文件名HBA_MOUSE.FASTA后系统提示输入起始空位罚分值Gap opening penalty,并给出默认值10.0,按回车键Enter则采用默认值。
5) 系统提示输入延伸空位罚分值Gap extension penalty,并给出默认值0.5,按回车键采用默认值。
6) 系统提示比对结果输出文件名Output alignment,并将第一个序列FASTA格式第1行注释信息中的序列名作为默认输出结果文件名hba_human.needle(默认输出文件名用小写字母)。也可由用户输入指定的文件名,如此处的HUMAN-MOUSE.NEEDLE。
1.3.2 参数式上述交互式运行模式适用于初学者,用户可根据提示信息确定需要输入的文件名和参数,许多情况下可先使用默认值,在分析运行结果后再次运行时则可适当调节参数,以得到更好的结果。对于熟练用户,则可采用参数模式运行程序,即在命令行中直接给出输入文件名、输出文件名和程序参数。例如,用needle程序对人、小鼠和大鼠三种哺乳动物alpha血红蛋白进行两两比对,则可分别采用以下命令。

上述needle程序运行过程说明如下。
1) 第一次对人和小鼠alpha血红蛋白进行比对,采用默认起始空位罚分-gapo 10.0和默认延伸空位罚分-gape 0.5,运行结果存放到输出文件HUMAN-MOUSE.NEEDLE中。参数-gapo是-gapopen的简略形式,-gape是-gapextension的简略形式。
2) 第二次对人和大鼠alpha血红蛋白进行比对,空位罚分参数同上,运行结果存放到输出文件HUMAN-RAT.NEEDLE中。
3) 第三次对小鼠和大鼠alpha血红蛋白进行比对,空位罚分参数同上,运行结果存放到输出文件MOUSE-RAT.NEEDLE中。
上述命令的运行结果包括比对分值、相同位点数及百分比、相同及相似位点数及百分比(见表 1)。
表1(Table 1)
HUMAN-MOUSE.NEEDLE 648 122/142 (85.9) 131/142 (92.3)
HUMAN-RAT.NEEDLE 587 111/142 (78.2) 120/142 (84.5)
MOUSE-RAT.NEEDLE 632 120/142 (84.5) 127/142 (89.4)
表 1 人、小鼠和大鼠alpha血红蛋白两两比对结果Table 1 Results of pairwise sequence alignment of alpha hemoglobin between human, mouse, and rat
采用参数式运行模式时,输入文件、输出文件和程序参数均在命令行中给出,运行过程中不必逐个输入;与交互模式相比,运行效率有所提高,特别适合批量处理。
1.3.3 菜单式采用交互式运行时,除个别参数可由用户输入外,大部分参数由系统默认给定。若用户需要改变这些默认参数,则可采用菜单式运行。即在输入程序名时,同时输入选项参数-options,程序运行时则列出所有可选参数。这种运行方式,对具有较多选择参数的程序十分便利。下面,我们以豌豆开花后特异表达基因(Pea post-floral-specific gene, PPF-1)为例,说明如何按菜单式运行程序getorf,从mRNA中提取编码区(Coding sequence, CDS)序列。
1997年,朱玉贤等利用差异表达方法,从豌豆天然突变体G2株系中分离到开花后特异表达基因。为探索该基因是否与衰老相关,对其进行了序列分析,初步推断该基因的表达产物为内膜蛋白(inner-membrane protein),定位于叶绿体中,与某些细菌内膜蛋白有相同的亲疏水性模式[7]。
上述豌豆开花后特异表达基因序列分析的第一步,需要提取mRNA序列中自起始密码子ATG到终止密码子之间的编码区序列。该序列已递交NCBI核酸序列数据库GenBank(登录号Y12618),并作了初步注释,序列全长1 523个核苷酸,包括第48-1 373位编码区、5’端和3’端非翻译区(Untranslated region, UTR)。
从核酸序列数据库GenBank中下载FASTA格式序列文件Y12618.FASTA,采用菜单式运行编码区提取程序getorf,步骤如下。

上述运行过程说明如下。
1) 输入程序名并启用菜单式选项getorf -option。
2) 输入豌豆开花后特异表达基因mRNA序列Y12618.FASTA。
3) 系统显示0-23种可选遗传密码表,按回车键选择默认通用密码表。
4) 系统显示最小读码框长度,默认为30,输入1 000,仅获取长度大于1 000 bp的编码区序列。
5) 系统显示最大读码框长度,按回车键选择默认值1 000 000。
6) 系统显示输出序列种类,共有7种不同选择,输入1则提取编码区序列并翻译成氨基酸。
1.4 三个帮助程序EMBOSS软件包整合了300多个程序,可通过三个帮助程序wossname, tfm和seealso了解某个程序的用途和用法。
1.4.1 wossname第一个程序为wossname,其含义为What’s the name,可通过输入关键词查找特定用途的程序名称。例如,可用以下命令找到所有点阵图(Dot-plot)序列比对程序。

1.4.2 tfm第二个帮助程序为tfm,其含义为The file manual,可用来显示某个程序的使用方法和可选参数,内容十分详尽,命令参数部分列出可供用户选择的所有参数及其数据类型,并给出默认值和可选范围(见表 2)。
表2(Table 2)
Wiki EMBOSS网站在线手册
Function 程序功能和用途
Description 程序简单描述
Algorithm 算法
Usage 使用方法和实例
Command Line Arguments 命令行参数
Input File Format 输入文件格式及实例
Output File Format 输出文件格式
Data Files 参数文件(如计分矩阵、密码子使用表等)
References 参考文献
See Also 与本程序相关的其它程序
Author 程序编写者
History 程序编写时间和修改历史
Target Users 目标用户
Comments 注解
表 2 EMBOSS软件包中tfm程序帮助信息Table 2 Help information of the tfm program in EMBOSS
命令tfm的功能与Linux系统man命令的功能类似,详细列出某程序所有帮助信息。此外,也可以在程序运行时用-help参数列出该程序简略信息,包括EMBOSS软件包的版本。
1.4.3 seealso第三个帮助程序为seealso,即英文See also,其含义为列出EMBOSS软件包中与某程序相关的其它程序。例如,以下命令列出与点阵图程序dotmatcher相关的其它点阵图程序。

2 序列处理EMBOSS软件包整合的程序几乎涵盖了序列分析的所有方面。本文按功能分类,简要介绍部分常用程序,包括序列处理程序、序列比对程序、核酸序列分析程序和蛋白质序列分析程序,对其中一些具有代表性的程序结合实例给出具体操作步骤和运行结果,并对运行结果的生物学意义略加说明。
序列处理是序列分析的基础,下面我们分别介绍最为常用的格式转换、序列提取和序列变换三类序列处理程序。
2.1 格式转换核酸和蛋白质序列格式有多种,不同格式之间的转换在序列分析中经常遇到。EMBOSS程序多以FASTA作为输入序列格式。从GenBank, EMBL等核酸序列数据库和UniProt等蛋白质序列数据库下载的原始格式序列条目,可转换成FASTA格式。
EMBOSS包括多个序列格式转换程序,此处介绍最为常用的seqret。该程序是EMBOSS软件包开发的第一个程序,除了格式转换外,还有其它许多功能。
2.1.1 seqret用法实例从NCBI核酸序列数据库下载GenBank格式豌豆开花后特异表达基因mRNA序列(登录号Y12618),以Y12618.GB为文件名保存。可用seqret程序将其转换成FASTA格式。采用参数式方法,直接在命令行指定输入文件Y12618.GB和输出文件Y12618.FASTA。

上述豌豆开花后特异表达基因的表达产物为内膜蛋白,已在UniProt蛋白质数据库Swiss-Prot子库中收录。UniProt数据库中每个序列都有特定的序列条目名(Entry name)。下载UniProt/Swiss-Prot格式豌豆内膜蛋白序列(序列条目名PPF1_PEA),保存为PPF1_PEA.SW,可用以下命令转换成FASTA格式序列文件。

2.2 序列提取EMBOSS软件包中的序列提取程序,包括以下两大类。第一类用于FASTA格式输入文件。这类程序操作比较简单,常用的有seqretsplit和extractseq,前者可将一个FASTA格式多序列文件拆分成多个FASTA格式单序列文件,后者可根据用户指定的区域,提取序列中的子序列,合并成新序列,或按多个序列保存。
另一类程序则用于GenBank和RefSeq格式的核酸序列或UniProt格式的蛋白质序列。这些数据库中的序列条目通常均包含序列特征注释信息,也称序列特征表(Feature Table)。这里程序则可根据序列特征表中提供的注释信息,提取其中的子序列。核酸序列数据库的序列特征表包括mRNA、编码序列、翻译产物蛋白质、外显子(Exon)、内含子(Intron)、非翻译区、序列标签位点(Sequence Tag Site, STS)等。蛋白质序列数据库的序列特征表包括二级结构(HELIX, STRAND, TURN)、跨膜螺旋(TRANSMEM)、变异位点(VARIANT)、活性位点(ACT_SITE)、金属结合位点(METAL)、糖基化位点(CARBOHYDR)、DNA结合区域(DNA_BIND)、二硫键(DISULFID)、信号肽(SIGNAL)、序列模体(MOTIF)和结构域(DOMAIN)等[5]。
这类程序中最为常用的有coderet和extractfeat。coderet用法比较简单,可用于提取mRNA、编码序列和所编码的蛋白质序列。extractfeat功能十分强大,用法也比较灵活,可提取更多种类的子序列,包括外显子、内含子、重复序列和多聚腺苷酸信号等。此外,通过设定特征表类型(Type)、标签(Tag)和标签值(Value)等参数,也可提取用户指定的一个或几个特定子序列。
2.2.1 coderet用法实例以小鼠alpha血红蛋白编码基因(GenBank登录号V00714)DNA序列为例,根据序列注释信息,该基因包括三个外显子,其mRNA序列位于372-500, 623-826和961-1 191,编码区位于405-500, 623-826和961-1 089。运行coderet程序,可分别提取mRNA序列、编码区序列、翻译产物蛋白质序列和非编码区序列。

调用coderet程序,输入小鼠alpha血红蛋白GenBank格式文件V00714.GB,程序以GenBank序列条目中基因座(LOCUS)名称v00714为默认输出文件名,将输出结果分别保存到5个文件中,分别为列表文件(v00714.coderet)、编码区序列文件(v00714.cds)、mRNA序列文件(v00714.mrna)、所编码的蛋白质序列文件(v00714.prot)和非编码区序列文件(v00714.noncoding)。按EMBOSS软件包习惯,默认输出文件名用小写字母表示。
2.2.2 extractfeat用法实例以上述小鼠alpha血红蛋白编码基因为例,根据序列注释信息,该基因包括两个内含子,分别位于501-622和827-960,用以下命令可提取这两个内含子的序列:

2.3 序列变换EMBOSS软件包中的序列变换程序包括reveseq, msbar和shuffleseq等。程序revseq将已知序列转换成反向互补序列,msbar对已知序列进行突变,shuffleseq则用于产生随机序列。
程序msbar可用于对已知序列进行单点或多点随机突变,突变方式可以是替换、插入或删除,突变位点可以是单个核苷酸点突变(Point mutation),也可插入或删除一个序列片段(Block mutation),还可插入或删除一个密码子(Codon mutation)。程序运行默认方式为交互式,即屏幕显示交互菜单,用户可以自行选择突变次数(Number of times),也可按上述三种不同突变时,选择插入(Insertion)、删除(Deletion)、替换(Substitution)、复制(Duplication)和移动(Move)等不同突变方式。以豌豆开花后特异表达基因编码区序列为例,以下命令对该序列进行1次单点插入突变、1次片段删除突变和1次密码子替换突变。

突变结果可用双序列比对程序needle验证。由于程序msbar基于随机突变,即使运行时设定的参数完全一致,两次运行结果并不相同。
2.4 序列显示EMBOSS软件包中的序列显示程序包括infoseq、showseq和showfeat等。程序infoseq显示序列简单信息,包括序列名称、长度和GC含量等,showseq按不同格式输出序列,而showfeat则根据序列特征表输出序列注释信息。
2.4.1 infoseq用法实例程序infoseq简单实用,可用于显示序列名称、格式及登录号等基本信息,并可统计序列长度。对于核酸序列,还能统计GC含量。可用以下命令显示豌豆开花后特异表达基因的基本信息。

程序infoseq既可用于GenBank和Swiss-Prot等格式,也可用于FASTA格式;既可用于单个序列,也可用于多序列文件。可用以下命令显示12个人源癌胚抗原(Carcinoembryonic Antigen, CEA)蛋白质分子的基本信息[8]。

2.4.2 showseq用法实例程序showseq可用不同方式显示核酸序列,也可显示按不同读码框翻译得到的氨基酸序列、反向互补序列,以及酶切位点等。以下命令显示豌豆开花后特异表达基因第1-120位序列,并用标尺显示序列位点,第48-120位用大写字母显示。

2.4.3 showfeat用法实例程序showfeat可用于显示GenBank和Swiss-Prot等序列的特征信息。以下命令以图形方式显示GenBank格式小鼠alpha血红蛋白编码基因V00714.GB中外显子和内含子位置。

3 序列比对序列比对在生物信息学中占有重要地位,是核酸和蛋白质序列分析的基础。EMBOSS软件包整合了十多个序列比对程序,包括双序列比对、多序列比对、数据库搜索,以及基于点阵图的可视化序列比对等。
3.1 双序列比对EMBOSS中整合的双序列比对程序包括needle, water, stretcher, matcher, seqmatcherall, supermatcher和esim4等,其中needle和stretcher为基于全局相似性的序列比对程序,其余为基于局部相似性的序列比对程序。needle和water最为常用,广泛用于核酸和蛋白质序列比对。它们均基于动态规划算法,在给定计分矩阵和空位罚分前提下,能够得到最佳比对结果,即最优解。程序needle所采用的算法由Needleman和Wunsch于1970年提出,而程序water所采用的算法由Smith和Waterman于1981年提出。程序stretcher是在needle基础上稍作修改,运行时所需内存大为降低,而运行时间稍长。而程序matcher则是water的改进版,可由用户指定输出一个或多个最佳局部比对结果。程序seqmatcherall和supermatcher用于多条序列比对或数据库搜索,运行时间较长。此外,esim4是将mRNA序列定位于基因组序列的程序,而est2genome则是将表达序列标签(Expressed Sequence Tag, EST)定位于基因组序列的程序。
3.1.1 needle用法实例下面,我们以人癌胚抗原为例,说明全局比对程序needle和局部比对程序water的用途和用法。
人癌胚抗原是一种细胞表面糖蛋白,多在直肠癌、胃癌等恶性肿瘤中表达[8]。CEA基因家族分CEA和妊娠特异性beta-1糖蛋白(Pregnancy-specific beta-1-glycoprotein, PSG)两个亚家族,其中CEA亚家族包括12个不同成员(见表 3)。CEA蛋白质分子属免疫球蛋白超家族,N-端含长度为34个氨基酸的信号肽,第35位开始则为免疫球蛋白可变结构域(Immunoglobin Variable Domain, IgV),长度约为110个氨基酸。除可变结构域外,有的CEA分子还含一个或多个免疫球蛋白恒定结构域(Immunoglobin Constant Domain, IgC),分不同亚型。
表3(Table 3)
P40198 CEAM3_HUMAN 252 N TMH
O75871 CEAM4_HUMAN 244 N TMH
Q7Z692 CEA19_HUMAN 300 N TMH
Q14002 CEAM7_HUMAN 265 N-A GPI
Q3KPI0 CEA21_HUMAN 293 N-A TMH
Q2WEN9 CEA16_HUMAN 425 N1-A-B-N2 None
P40199 CEAM6_HUMAN 344 N-A-B GPI
P31997 CEAM8_HUMAN 349 N-A-B GPI
A8MTB9 CEA18_HUMAN 384 N-A-B TMH
P13688 CEAM1_HUMAN 526 N-A1-B-A2 TMH
Q6UY09 CEA20_HUMAN 596 n-A1-B1-A2-B2 TMH
P06731 CEAM5_HUMAN 702 N-A1-B1-A2-B2-A3-B3 GPI
表 3 UniProt/Swiss-Prot中收录的12个人源癌胚抗原蛋白质分子Table 3 Twelve human CEA proteins in UniProt/Swiss-Prot
UniProt/Swiss-Prot蛋白质数据库中收录了12个人源癌胚抗原蛋白质CEA(见图 1,根据德国Ludwig-Maximilians大学Zimmermann教授CEA网站改编)。图中标有N的结构域为免疫球蛋白可变结构域IgV;标有A和B的结构域为免疫球蛋白恒定结构域IgC,各分3种亚型(A1, A2, A3和B1, B2, B3)。嵌入磷脂双层膜的箭头表示糖基磷脂酰肌醇(Glycosylphosphatidylinositol, GPI)膜结合位点,穿过磷脂双层膜的螺旋表示跨膜螺旋(Transmembrane helix, TMH)。
图 1(Figure 1)
 图 1 UniProt/Swiss-Prot数据库中12个人源癌胚抗原蛋白质分子Figure 1 Twelve human CEA proteins in UniProt/Swiss-Prot
图 1 UniProt/Swiss-Prot数据库中12个人源癌胚抗原蛋白质分子Figure 1 Twelve human CEA proteins in UniProt/Swiss-Prot人的Ⅲ型(CEAM3_HUMAN)和Ⅳ型(CEAM4_HUMAN)CEA分子长度接近,各含1个可变结构域、1个跨膜螺旋区和1个膜内区。用全局比对程序needle对其进行序列比对,命令如下。

分析比对结果可以发现,这两个亚型的CEA分子具有较高的相似性,仅在C-端有1个长度为8个氨基酸残基的插入。上述命令中,起始空位罚分设为20,而不用默认值10,延伸空位罚分设为2,而不用默认值0.5,可用来避免不必要的插入或删除。
3.1.2 water用法实例全局比对程序needle适用于长度相差不大的两个序列,如上述CEAM3_HUMAN和CEAM4_HUMAN,而CEAM5_HUMAN含7个结构域,除可变结构域IgV外,还包括6个恒定结构域IgC。若需比较其可变结构域与CEAM3_HUMAN的可变结构域序列相似性,则可用局部比对程序water进行比对。这两个分子N-端均有长度为34个氨基酸的信号肽,C-端有跨膜螺旋,免疫球蛋白结构域位于35-142区域,比对时可用参数指定。

比对结果表明,这两个CEA分子N-端可变结构域序列具有较高相似性。
3.2 多序列比对和双序列比对一样,多序列比对在核酸和蛋白质序列分析中也广泛使用。EMBOSS软件包中整合了多序列比对程序emma和edialign。程序emma是EMBOSS整合的基于全局相似性多序列比对程序ClustalW,而edialign则是EMBOSS整合的基于全局加局部相似性的多序列比对程序Dialign。
3.2.1 emma用法实例下面以血红蛋白为例,说明emma用法。UniProt/Swiss-Prot中收录了9个已经审阅的人源血红蛋白[4],分属于alpha和beta两个亚家族。alpha亚家族有5个基因,编码4种蛋白质,其中HBA1和HBA2两个基因编码的蛋白质序列完全相同;beta亚家族也有5个基因,编码5种蛋白质(见表 4)。
表4(Table 4)
Accession 序列条目名
Entry name 基因名
Gene name 序列长度
Length 亚家族
Sub-family
P02008 HBAZ_HUMAN HBZ 142 Alpha
Q6B0K9 HBM_HUMAN HBM 141 Alpha
P69905 HBA_HUMAN HBA1; HBA2 142 Alpha
P09105 HBAT_HUMAN HBQ1 142 Alpha
P68871 HBB_HUMAN HBB 147 Beta
P02042 HBD_HUMAN HBD 147 Beta
P69891 HBG1_HUMAN HBG1 147 Beta
P69892 HBG2_HUMAN HBG2 147 Beta
P02100 HBE_HUMAN HBE1 147 Beta
表 4 UniProt/Swiss-Prot中收录的人的9种不同血红蛋白Table 4 Nine human hemoglobins in UniProt/Swiss-Prot
可用以下命令对上述9个血红蛋白进行多序列比对,除输出FASTA格式序列比对文件外,同时输出Newick格式分支图文件,可用MEGA软件显示其树形分支结构。

程序emma有许多可调参数,包括计分矩阵、空位罚分、比对方式和输出格式等,可用菜单运行模式,即运行时加-options参数,即可指定上述参数的值。
3.2.2 edialign用法实例程序emma多用于全局比对,如上述9个长度相差不大的血红蛋白,而edialign采用全局比对加局部比对的方法,适用于寻找蛋白质序列中具有局部相似性的保守结构域或核酸序列中保守序列模体(Motif)。例如,12个人源癌胚抗原可用edialign进行多序列比对。

输出结果保存到两个文件中,12HUMAN_CEA.EDIA是多序列比对格式,比对结果中保守区域用大写字母表示,每个位点标有数字0-9,数字越大,保守性越高。12HUMAN_CEA.ALN为FASTA格式的比对结果。
3.3 点阵图点阵图也是序列比对中常用方法,其特点是输出结果直观。EMBOSS中整合了4个点阵图程序,即dottup, dotpath, dotmatcher和polydot。程序polydot用于多序列比对,而其余3个程序用于双序列比对。运行点阵图程序时,通常需要指定滑动窗口大小,若滑动窗口中两个序列片段相似性超过用户指定的阈值,则在平面坐标系中用点标出。需要注意的是,dottup和dotpath只考虑指定大小的滑动窗口中两个序列片段中相同核苷酸或氨基酸,可用于核酸或蛋白质序列比对;而dotmatcher不仅考虑相同残基,同时根据计分矩阵考虑氨基酸残基之间的相似性,只能用于蛋白质序列比对。
3.3.1 dottup用法实例下面,以河豚鱼质粒片段DNA序列为例,说明dottup的用法。人的多药耐药(Multidrug Resistance, MDR)基因家族包括MDR1, MDR3等几种不同亚型,其表达产物为膜通道糖蛋白,利用ATP提供能量,将药物等细胞内外源物质运送到胞外从而产生抗药性。为探索MDR耐药机制,刘勇于1998年从模式生物河豚鱼(Takifugu rubripes, Fugu)柯氏质粒(Cosmid)中克隆到两个人的MDR同源基因(补充材料)。这两个河豚鱼MDR基因头尾相接串联排列,测序拼接得到的全长序列约为40 kb。该河豚鱼序列片段已提交NCBI核酸序列数据库GenBank(登录号AF164138)。下载FASTA格式的河豚鱼DNA序列片段,利用点阵图程序dottup,可以输出图形文件,显示这两个基因的大体位置。
EMBOSS软件包中dottup等程序运行图形文件格式可由用户选择,缺省为X11,若装有图形显示终端(X-Terminal),可直接在屏幕上输出。也可保存为其它格式的图形文件,如可缩放矢量图形格式(Scalable Vector Graphics, SVG)和可移植网络图形格式(Portable Network Graphics, PNG)。
以下是利用EMBOSS软件包中点阵图程序dottup分别对河豚鱼基因组序列片段进行比对的命令和所用参数。

上述命令输出结果显示两条与对角线平行的线段(见图 2a),表明该基因组序列片段5’端有两个长度约为13kb相似片段,即两个串联重复多药耐药基因MDR。
图 2(Figure 2)
 图 2 河豚鱼多药耐药基因点阵图序列比对结果Figure 2 Dot-plot alignment output of Fugu multidrug resistance gene
图 2 河豚鱼多药耐药基因点阵图序列比对结果Figure 2 Dot-plot alignment output of Fugu multidrug resistance gene用以下命令,设定比对范围,则可进一步确定这两个串联重复基因的相似性。

从输出结果(见图 2b)中可以看出,这两个序列片段具有一定相似性,有些区域相似性较高,图中为连接在一起的线段,而有些区域相似性较低,可能是基因中内含子区域。
查看该基因组序列注释信息,发现这两个基因由20多个外显子组成。利用coderet程序,提取编码序列,运行dottup程序,比较这两个编码序列的相似性。

输出结果(见图 2c)显示,编码区序列相似性比全长基因序列相似性更高。运行dottup程序,可进一步比较所编码蛋白质序列相似性。

输出结果(见图 2d)显示,所编码两个蛋白质序列同样具有较高相似性。
3.3.2 Dotmatcher用法实例点阵图程序dottup多用于核酸序列,而dotmatcher则可用于蛋白质序列。下面以果蝇体节发育相关基因为例,说明利用dotmatcher显示序列中的重复片段。该基因编码长度为1 504个氨基酸的蛋白质(UniProt序列条目名SLIT_DROME)。可用以下dotmatcher命令和参数,得到不同输出结果。

查看数据库中SLIT_DROME序列特征表注释信息,其N末端有4个区域富含亮氨酸重复片段(Leucine Rich Repeat, LRR),每个区域由6个重复片段组成,每个重复片段约含24个氨基酸残基,序列上有一定保守性;C末端含7个类表皮生长因子(Epidermal Growth Factor like, EGF-like)结构域,每个结构域约含38个氨基酸残基。运行程序dotmatcher对其自身进行比对,采用不同大小的滑动窗口和相似性阈值,可得到不同结果(见图 3)。当窗口大小和相似性阈值均为默认值时,背景噪声较大(见图 3a);当把窗口大小改为24个残基,相似性阈值改为20时,可清晰显示长度为24的亮氨酸重复片段(见图 3b)。当窗口大小与重复单元大小相近时,所显示的重复区域最为清晰。当把窗口大小改为38个残基,相似性阈值改为20时,可清晰显示表皮生长因子结构域(见图 3c)。当保持窗口大小为38而相似性阈值改为30时,可减少背景噪声(见图 3d)。阈值越大,背景噪声越小。
图 3(Figure 3)
 图 3 果蝇体节发育基因蛋白质序列点阵图分析结果Figure 3 Dot-plot results of repeat regions in protein sequence of fruitfly midline development related gene
图 3 果蝇体节发育基因蛋白质序列点阵图分析结果Figure 3 Dot-plot results of repeat regions in protein sequence of fruitfly midline development related gene利用点阵图程序,通过设置适当参数,可清晰显示串联重复基因和重复结构域等。重复结构域在蛋白质分子中较为多见,如钙结合蛋白(CALB1_HUMAN)含5个长度为36 aa的EF-手型(EF-hand)重复单元,膜联蛋白A1(ANXA1_HUMAN)含4个长度为72-73 aa的膜联蛋白重复单元,抗肌萎缩蛋白(DMD_HUMAN)含24个长度为100-110 aa的红膜肽(Spectrin)重复单元。而肌联蛋白(TITIN_HUMAN)则是由多个不同重复单元组成的巨型蛋白质,全长34 350个氨基酸,含152个长度为90 aa左右的免疫球蛋白结构域(Ig-like)、132个长度为95 aa左右的Ⅲ型纤联蛋白(Fibronectin type-Ⅱ)、19个长度为45 aa左右的Kelch重复单元、17个长度为55 aa左右的RCC1重复单元、15个长度为40 aa左右的WD重复单元和14个长度为34 aa左右的TPR重复单元。
4 核酸序列分析4.1 序列组分统计组分分析是序列分析中最基本的方法之一。EMBOSS中用于核酸序列组分分析的程序包括geecee, freak, wordcount和compseq等,其中geecee和freak主要用于GC含量分析,wordcount和compseq用于统计四种核苷酸出现频率。
4.1.1 compseq用法实例程序compseq可用于按指定长度统计核酸序列中不同字串出现频率。所谓字串,是指一定长度的不同核苷酸组合。长度为2的2字串共有16种,即AA, AC, AG, AT, ……, TA, TC, TG, TT;长度为3的3字串有64种,即AAA, AAC, AAG, AAT, ACA, ACC, ACG, ACT, ……, TTA, TTC, TTG, TTT;4字串、5字串和6字串分别有256、1, 024和4, 096种。下面以三种模式微生物为例,分别统计6字串出现的次数和与期望值的比例。

上述程序运行结果每个序列都生成4 096个不同的6字串,其中有的6字串频数很低,有的6字串频数很高(见表 5)。这三种模式微生物在细菌基因组学研究中具有重要地位。大肠杆菌是人类基因组计划指定的模式微生物,结核分枝杆菌是最早完成基因组测序的致病菌,泉生热胞菌是我国科学家于2002年完成基因组测序的第一个细菌。
表5(Table 5)
学名 Escherichia coli Mycobacterium tuberculosis Caldanaerobacter subterraneus
菌株 K12/MG1655 CDC1551/H37RV tengcongensis MB4
登录号 NC_000913.3 NC_000962.3 NC_003869.1
文件名 ECOLI_K12.FASTA MYCTO_H37.FASTA CALSU_MB4.FASTA
长度 4 641 652 4 411 532 2 689 445
GC含量 50.79 65.61 37.57
低频串1 CCTAGG (16/0.014) ATTATA (10/0.009) CGCACG (23/0.035)
低频串2 CTAGGA (17/0.015) TATAAT (10/0.009) CGCGCG (24/0.037)
低频串3 TCTAGG (19/0.017) TATAAA (15/0.014) CGACCG (26/0.040)
低频串4 CTAGAC (20/0.018) TTATAA (20/0.019) CGATCG (29/0.044)
低频串5 TCCTAG (21/0.019) TATTAT (21/0.019) CGGTCG (31/0.047)
高频串1 CGCCAG (5 397/4.763) CCGCCG (11 203/10.402) AAAAAA (5 634/8.581)
高频串2 CTGGCG (5 267/4.648) CGGCGG (10 991/10.205) TTTTTT (5 528/8.419)
高频串3 GCCAGC (4 832/4.264) GCCGCC (10 238/9.506) AAAAAT (5 381/8.195)
高频串4 GCTGGC (4 765/4.205) GCCGGC (10 136/9.411) TAAAAA (5 378/8.191)
高频串5 CCAGCG (4 611/4.069) GGCGGC (9 929/9.219) ATTTTT (5 375/8.186)
表 5 三种模式微生物基因组序列中的特殊6字串Table 5 Special 6-mer in three models of bacterial genome sequences
4.1.2 freak用法实例程序compseq用于统计核酸序列中不同字串出现频率,而程序freak则可以图形方式输出不同区域GC含量。以下命令可显示小鼠alpha血红蛋白基因DNA序列(全长1 441 bp)不同区域GC含量分布。

从程序freak输出结果可以看出,小鼠alpha血红蛋白基因5’端和3’端GC含量较低,而在600-1 000 bp区域GC含量较高(见图 4)。
图 4(Figure 4)
 图 4 小鼠alpha血红蛋白基因不同区域GC含量Figure 4 GC content of different regions in mouse alpha hemoglobin gene
图 4 小鼠alpha血红蛋白基因不同区域GC含量Figure 4 GC content of different regions in mouse alpha hemoglobin gene4.2 开放读码框分析EMBOSS中整合的开放读码框分析程序包括plotorf, sixpack, showorf和getorf。这四个程序可以组合使用,plotorf用图形方式输出DNA序列中所有可能的读码框,即起始密码子和终止密码子之间、终止密码子和终止密码子之间的序列,包括三条正链和三条负链;sixpack给出DNA序列所有可能的编码方式;showorf按指定编码链输出DNA序列及其编码的氨基酸序列;getorf用于提取读码框序列或所编码的氨基酸序列,也可提取编码区上游或下游非翻译区序列。
本文1.3.3中介绍了getorf的用法,下面介绍sixpack和showorf的用法。
4.2.1 sixpack用法实例以豌豆开花后特异表达基因全长mRNA序列(GenBank登录号Y12618)为例,调用EMBOSS软件包中的sixpack程序,输出该序列正链(F1-F3)和互补链(F4-F6)序列,以及6条开放读码框所编码的氨基酸。

运行结果显示,正链第3条读码框(F3)第48位有起始密码子ATG,第1 374位和1 377位有两个连续终止密码子TGA和TAG,表明该序列编码区位于正链48-1 373位,共1 326 bp,编码442个氨基酸。
4.2.2 showorf用法实例上述以豌豆开花后特异表达基因全长mRNA序列为例,调用EMBOSS中showorf程序,指定第3条读码框,则输出该读码框序列及对应的氨基酸序列。

4.3 CpG岛识别CpG岛是指DNA序列中富含CG双核苷酸的区域,其顺序为C在前,G在后。为避免误解,常用CpG表示,即胞嘧啶3’端与鸟嘌呤5’端通过磷酸基团连接。CpG岛通常位于基因上游启动子区域300-3 000 bp区域内,该区域的特征是核苷酸G和C含量较高,且富含CpG双核苷酸。因此,CpG岛预测结果可用来推断某个DNA序列片段中是否存在蛋白质编码基因。
EMBOSS中整合的CpG岛分析程序包括cpgplot和cpgreport等。程序cpgplot用于预测DNA序列中的CpG岛,并以图形方式输出结果。程序cpgreport用于计算DNA序列中CpG双核苷酸含量,所用方法与cpgplot有所不同,灵敏度较高,但假阳性率也较高。
4.3.1 cpgplot用法实例人alpha血红蛋白基因家族分布在16号染色体短臂靠近端粒处,包括5个功能基因(zeta, mu, alpah2, alpha1和theta)以及两个假基因(HBZP和HBA1P)。该基因家族DNA序列长度为43 058 bp(GenBank登录号Z84721)。下载FASTA格式序列并运行cpgplot程序。
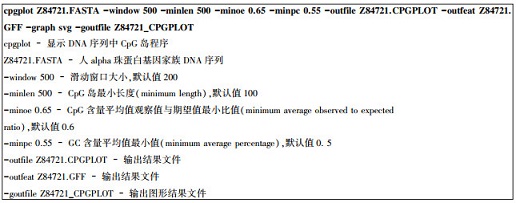
上述运行过程中,滑动窗口大小和CpG岛长度均设为500 bp,双核苷酸CpG含量观察值与期望值比值下限设为0.65,GC含量下限设为0.55。查看该序列条目Z84721中注释信息,预测结果与注释信息比较吻合。若采用系统给定缺省参数,预测灵敏度较高,但假阳性率也较高。结果表明,该基因组序列片段中有5个CpG岛(见表 6)。
表6(Table 6)
HBAZ 14 618-15 730 1 112 14 606-15 663 1 058 73.82
HBM 25 208-27 584 2 376 25 196-27 543 2 348 72.44
HBA2 33 234-34 311 1 077 33 080-34 404 1 325 70.87
HBA1 37 038-38 118 1 080 36 880-38 206 1 327 70.99
HBQ1 41 026-42 625 1 599 40 933-42 636 1 704 69.72
表 6 人alpha血红蛋白基因家族序列中的CpG岛Table 6 CpG island of human alpha hemoglobin gene cluster
程序cpgplot除了输出文本文件Z84721.CPGPLOT外,还可输出图形文件,以波形图方式显示序列不同区域CpG双核苷酸的含量和可能的CpG岛位置(见图 5)。
图 5(Figure 5)
 图 5 程序cpgplot分析人alpha血红蛋白基因簇序列中的CpG岛输出结果Figure 5 Cpgplot result of human alpha hemoglobin gene cluster
图 5 程序cpgplot分析人alpha血红蛋白基因簇序列中的CpG岛输出结果Figure 5 Cpgplot result of human alpha hemoglobin gene cluster4.4 密码子分析密码子一共有64个。密码子具有通用性、简并性和偏好性等特点。除个别特殊密码子外,通用密码子中ATG为起始密码子或编码甲硫氨酸(Met, M);TAA, TAG和TGA为终止密码子;其余60个密码子编码19种氨基酸。编码同一氨基酸的密码子使用频率可能不同,即密码子使用具有偏好性(Codon Usage Bias)。分析密码子使用频率及偏好性,是系统发育和分子演化研究的常用方法。
EMBOSS中密码子分析程序包括cusp、syco、cai和chips等。其中cusp用于统计核酸序列中64种密码子使用频率和期望值,并给出编码同一氨基酸的不同密码子使用比例。syco用图形方式显示同义密码子使用偏好,可用于基因预测。cai用于计算密码子适应指数(Codon Adaptation Index),取值范围为0-1;cai值越大,密码子使用偏好性越强。一般说来,表达量高的基因,其密码子使用偏好性强;因此,cai值可用于预测基因表达水平高低。chips用于计算有效密码子数(Effective Number of Codon, ENC),其范围为20-61。ENC值越小,密码子使用偏好性越强。不同物种或同一物种的不同基因,其ENC值有所不同。
4.4.1 cusp用法实例以豌豆开花后特异表达基因Y12618编码区序列为例,程序cusp运行结果为不同密码子使用频率。结果表明,该编码区序列密码子第3位偏好使用A或T。

4.4.2 chips用法实例以豌豆开花后特异表达基因Y12618编码区序列为例,运行chips程序,可得到有效密码子ENC值。

4.5 重复序列寻找重复序列在核酸序列中十分普遍。常见的重复序列包括串联重复(Tandem Repeat)和倒转重复(Inverted Repeat)。串联重复是指某一特定序列片段连续多次重复排列,如DNA序列中微卫星(Microsatellite)序列、短散在重复元件(Short Interspersed Nuclear Elements, SINE)和长散在重复元件(Long Interspersed Nuclear Elements, LINE)等。倒转重复是指同一条链上两个序列片段通过碱基配对反向互补,如microRNA前体序列中的茎环结构(Stem-loop)。
EMBOSS中重复序列分析程序包括etandam、equicktandem、einverted和palindrome等。程序etandem和equicktandem用于寻找核酸序列中串联重复序列,前者允许空位插入,而后者不允许插入空位,运算速度较快。程序palindrome和einverted用于寻找倒转重复序列,前者用于寻找较短片段的回文结构,两个配对序列之间可以有错配,但不允许空位插入;而后者用于寻找较长的倒转重复,既可以有错配,也可以有空位插入。
4.5.1 palindrome用法实例以拟南芥microRNA 172c(ath-MIR172c)为例,从microRNA数据库下载前体序列(登录号MI0000991),利用seqret程序将其转换成FASTA格式,并将字符U替换成T。运行程序palindrome,用交互式方式设置参数:最小反向重复序列长度22,最大反向重复序列长度25,反向重复序列间最大间隔100,允许错配核苷酸数2,输出结果包括互相重叠的重复序列。

运行结果表明,microRNA前体序列ath-MIR 172c中17-38/96-117位为倒转重复序列。查看microRNA数据库miRBase中的注释信息,该前体序列成熟miRNA位于98-118位。
4.5.2 einverted用法实例以果蝇性别相关基因为例,从GenBank下载序列(登录号EF565211),运行程序einverted,参数设置采用系统默认值,空位罚分12、最小分值50、匹配分值3和错配分值-4。输出结果为倒转重复文件和FASTA格式序列文件。

运行结果表明,该序列中1 617-1 966/2 355-2 699位为倒转重复,中间有1个长度为6的空位。查看该序列注释信息,该倒转重复序列与果蝇性别比例抑制功能有关。
5 蛋白质序列分析5.1 序列组分统计EMBOSS中用于蛋白质一级结构氨基酸序列统计分析的程序包括pepstats, pepinfor, wordcount和compseq等。pepstats以文本和表格方式输出蛋白质序列中各种氨基酸含量,并对不同类型氨基酸进行统计,如亲水氨基酸和带电氨基酸等;pepinfor则以图形方式显示各种类别氨基酸在序列不同区域的分布,如疏水性氨基酸、极性氨基酸、带电氨基酸和芳香族氨基酸等。此外,用于核酸序列组分分析的wordcount和compseq也可用于蛋白质序列组分分析。
5.1.1 pepstats用法实例以水稻落粒控制基因sh4蛋白质产物(UniProt序列条目Q1PIH9_ORYSI)为例,运行程序pepstats,则可统计20种氨基酸出现频率。

运行结果输出20种氨基酸频数和百分比。水稻落粒控制基因所编码蛋白质长度为390个氨基酸残基,不同氨基酸出现频率很不均匀,某些氨基酸出现频率较高,如脯氨酸、丙氨酸、丝氨酸和精氨酸高于平均值,而苯丙氨酸、异亮氨酸和甲硫氨酸则低于平均值。
5.1.2 Wordcount用法实例从以上分析可以看出,水稻落粒控制基因编码蛋白质中有些氨基酸出现频率偏高。利用程序wordcount,指定不同字长,可进一步分析水稻落粒控制基因蛋白质产物短片段重复序列出现频率。

结果发现,该蛋白质序列中存在大量短片段重复序列(见表 7)。
表7(Table 7)
频率 序列 频率 序列 频率 序列
13 PPP 7 PPPP 5 PPPPP
6 AAA 3 PPPS, AAAA 2 PPPPS, PPPSP, HGHGH, GGAAA, MSGSS,
4 PPS, HGH, HHH, GGA, EEE 3 PPSP, GGAA, GAAA, APPP, PLAL, HHHH, SGSS, HGHG, GHGH, MSGS
表 7 水稻落粒控制基因所编码的蛋白质序列中短肽重复片段Table 7 Short peptide repeats in protein sequences of rice shattering control gene
5.2 序列特征位点识别EMBOSS中用于蛋白质序列特征位点分析的程序包括antigenic、sigcleave和digest等,其中antigenic用于抗原决定簇分析,sigcleave用于信号肽剪切位点分析,digest用于酶切位点分析。
5.2.1 antigenic用法实例人III型癌胚抗原(CEAM3_HUMAN)胞外区35-142位为免疫球蛋白可变结构域。利用antigenic程序,可预测该结构域抗原决定簇,即可能的抗体结合部位。

预测结果表明,该蛋白质分子可能有三个抗原决定簇,分别位于第48-66, 75-86和119-129位。
5.2.2 fuzzprot用法实例程序fuzzprot用于寻找蛋白质序列中的序列模体。下面我们用植物特异转录因子家族Squamos promoter binding protein(SBP)为例,说明fuzzprot的用法。
植物转录因子数据库收录了17个拟南芥SBP家族基因,共30种不同转录本,编码17个转录因子。这17个转录因子的DNA结合结构域长度为79个氨基酸(见图 6),含两个锌指结构(Zinc finger)和1个核定位信号(Nuclear localization signal, NLS)。该核定位信号的序列比较保守,富含带正电的精氨酸(Arg, R)和赖氨酸(Lys, K)。利用以下命令可以找出核定位信号在17个转录因子DNA结合结构域中的位置。

图 6(Figure 6)
 图 6 拟南芥17个植物特异转录因子SBP家族DNA结合结构域序列图标Figure 6 Sequence logo of DNA binding domain in 17 SBP plant-specific transcription factors
图 6 拟南芥17个植物特异转录因子SBP家族DNA结合结构域序列图标Figure 6 Sequence logo of DNA binding domain in 17 SBP plant-specific transcription factors序列模体由用户指定,保守氨基酸用大写字符表示,中括号内为可选氨基酸,x为任意氨基酸,括号中为任意氨基酸个数,此处为6(输入括号时需要加转义符反斜杠,否则无法正常运行)。
5.3 二级结构分析EMBOSS中用于蛋白质二级结构分析的程序包括tmap, topo, pepwheel, helixturnhelix, pepcoil和garnier等,其中tmap用于跨膜螺旋预测,topo用于显示跨膜螺旋拓扑结构,helixturnhelix用于预测螺旋-转角-螺旋构象,pepcoil用于预测无规卷曲肽段,pepwheel用于显示alpha螺旋轮,garnier用于二级结构预测。
5.3.1 tmap用法实例以豌豆内膜蛋白(UniProt序列条目PPF1_PEA)为例,用以下命令运行程序tmap,可预测跨膜螺旋:

预测结果同时以文本格式和图形格式输出。结果表明,豌豆内膜蛋白序列中有4个可能的跨膜螺旋。跨膜螺旋长度为20-22个氨基酸,通常疏水性氨基酸为主。提取其中的第1个跨膜螺旋序列,保存为FASTA格式序列文件PPF1_PEA_H1.FASTA,可用pepwheel程序绘制alpha螺旋轮。
5.3.2 pepwheel用法实例对上述tmap程序预测所得第1个alpha螺旋FASTA格式序列,用以下命令运行pepwheel可绘制alpha螺旋轮。结果表明,该跨膜螺旋轮主要由疏水氨基酸组成。

5.3.3 garnier用法实例以抹香鲸肌红蛋白(PDB登录号1MBN)、人癌胚抗原N-端结构域(PDB登录号2QSQ)和人泛素蛋白(PDB登录号1UBQ)为例,从PDB数据库分别下载这3个蛋白质分子FASTA格式序列,分别用以下命令运行程序garnier,即可预测得到二级结构。

预测结果中字母H表示alpha螺旋(Helix),E表示beta折叠(Extended)、T表示转角(Turn)、C表示无规卷曲(Coil);下划线表示预测准确区域。这3种蛋白质分子的三维空间结构均已由实验测定,利用蛋白质结构显示和分析软件可观察这3种蛋白质分子的实际构象。肌红蛋白全长153个氨基酸残基,由8股alpha螺旋组成,按N-端到C-端顺序编号为A-H;癌胚抗原N-端结构域全长110个氨基酸残基,由9个beta折叠片组成,按N-端到C-端顺序为A-G(C和D之间另有C’和C”两个beta折叠片)。泛素蛋白全长76个氨基酸残基,由5个beta折叠(A, B, D, E, G)和2个alpha螺旋(C, F)组成。与实际构象比较表明,预测结果有一定误差。二级结构预测方法很多,目前预测精度为70%-80%。除EMBOSS中整合的garnier外,许多网站提供在线预测工具。读者可尝试不同程序,比较预测结果。
6 总结和讨论6.1 使用说明以上我们介绍了EMBOSS软件包中一些常用程序。经过十年多开发,EMBOSS已成为核酸和蛋白质序列分析常用软件包,为广大生物学工作者广泛使用。EMBOSS软件包功能齐全、用途广泛。选修“实用生物信息技术”课程的同学,在学习EMBOSS软件包中常用程序后,编写了以下顺口溜。
EMBOSS软件包,包罗万象真的好,
核酸蛋白都适用,功能强大效率高。
比对进化设引物,翻译酶切找重复,
程序名称不记得,wossname帮你找。
程序命令不会用,tfm把你教。
参数设置技巧高,点点滴滴要记牢,
EMBOSS是法宝,活学活用不愁了。
要熟练使用EMBOSS软件包中的程序,首先必须熟悉分子生物学基本概念,如中心法则和序列-结构-功能关系等;掌握必要的分子生物学基础知识,如基因结构、启动子、外显子、内含子、编码序列、RNA二级结构、蛋白质结构层次、一级结构序列特征和二级结构单元,以及序列模体、结构域、蛋白质家族和蛋白质功能等。
选择合适的程序、设置恰当的参数,是正确、高效使用EMBOSS软件包的关键。除了深入了解所研究问题的生物学背景外,也应搞清输入数据的种类、格式,掌握各种参数的含义,对所用程序的算法有所了解。同样一个问题,使用不同程序,运行结果就可能不同;即使是同一个程序,参数不同,结果也可能不同。熟练使用EMBOSS软件包提供的三个帮助程序wossname、tfm和seealso,深入理解各个程序的功能和可供设置的参数,可以在程序使用过程中起到事半功倍的效果。
需要说明的是,EMBOSS软件包启动时,人类基因组计划尚未完成,基因组数据分析刚刚开始。因此,EMBOSS不是组学数据分析软件,而是针对单个或多个蛋白质或核酸序列分析工具,其功能相当于基于个人计算机的共享软件BioEdit或商业软件DNAStar和MacVector等。从事基因组和转录组等高通量数据分析的读者,可选择Bowtie, BWA, TopHat/Cufflinks等软件。此外,EMBOSS软件包是单个程序的集成,各个程序之间并无联系,而后来开发的Galaxy平台,则将某些工具整合而形成互相关联的分析流程。
与所有计算机软件均可能存在“bug”一样,EMBOSS软件包中某些程序在运行时结果可能有误。例如,点阵图程序dotmatcher和dottup在比较两个不同序列时,坐标轴显示有误。此外,由于近年来UniProt数据库格式有所调整,序列提取程序extractfeat在处理UniProt/Swiss-Prot格式输入文件时,得不到正确结果。读者可自行修改源代码改正错误,或与Peter Rice联系。
6.2 程序列表2016年发布的EMBOSS 6.6.0版包括300多个程序,本文介绍的程序只是其中一小部分。为便于读者查询,我们按类别列出其中部分常用程序(见表 8)。
表8(Table 8)
seqret 格式转换 将GenBank, RefSeq, UniProt格式转换成FASTA格式
seqretsplit 序列提取 将一个FASTA格式多序列文件拆分成多个FASTA格式单序列文件
extractseq 序列提取 根据用户指定的区域,提取序列中的子序列
coderet 序列提取 根据序列特征表注释提取核酸序列中mRNA、编码序列和蛋白质序列
extractfeat 序列提取 根据序列特征表注释提取核酸或蛋白质序列中的子序列
revseq 序列变换 将输入序列转换成反向互补序列
msbar 序列变换 对输入序列进行模拟突变
shuffleseq 序列变换 对输入序列进行变换,产生随机序列
infoseq 序列显示 按表格方式显示序列长度、名称等基本信息
infoalign 序列显示 按表格方式显示多序列比对结果
showseq 序列显示 按一定格式显示核酸序列及特征信息
showpep 序列显示 按一定格式显示蛋白质序列及特征信息
showfeat 序列显示 按一定格式显示序列特征表信息
showalign 序列显示 按一定格式显示多序列比对结果
prettyseq 序列显示 用一定格式显示核酸序列
prettyplot 序列显示 用一定格式显示多序列比对结果
needle 序列比对 基于Needleman-Wunsch动态规划算法全局相似性双序列比
water 序列比对 基于Smith-Waterman动态规划算法局部相似性双序列比对
stretcher 序列比对 采用改进的Needleman-Wunsch算法双序列比对程序,占用内存较少,运行时间长
matcher 序列比对 基于局部相似性的双序列比对,通过设定参数,可同时输出多个相似性片段
supermatcher 序列比对 基于局部相似性双序列快速比对,适用于超长序列或与数据库之间相似性比对
seqmatcherall 序列比对 基于局部相似性双序列快速比对,用于寻找一组序列中所有匹配字串
esim4 序列比对 将mRNA序列定位于基因组序列
est2genome 序列比对 将EST序列定位于基因组序列
dottup 点阵图 显示两条序列之间相似性区域或一条序列中重复序列
dotpath 点阵图 显示两条序列之间主对角线相似性区域,不显示重复片段
dotmatcher 点阵图 显示两条蛋白质序列之间相似性区域或一条序列中重复序列片段
polydot 点阵图 显示多条序列之间相似性区域
compseq 组分分析 统计DNA或蛋白质序列中指定长度的各种组分观察频率和期望频率
wordcount 组分分析 统计DNA或蛋白质序列中指定长度的各种组分频数并以高低为序输出结果
geecee 组分分析 计算核酸序列中GC含量比例
freak 组分分析 以滑动窗口方式统计DNA序列中特定字串出现频率,用表格或图形方式输出结果
plotorf 读码框分析 根据起始密码子和终止密码子位置用图形方式显示mRNA序列开放读码框
sixpack 读码框分析 显示mRNA序列6个开放读码框和翻译所得氨基酸序列
showorf 读码框分析 显示mRNA序列指定读码框翻译所得蛋白质序列
getorf 读码框分析 从mRNA序列中提取开放读码框序列或其编码的氨基酸序列
cpgplot cpG岛分析 预测核酸序列中的CpG岛,用图形方式输出结果
cpgreport cpG岛分析 识别核酸序列中富含CpG双核苷酸区域
cusp 密码子分析 统计核酸序列各种密码子使用频率
syco 密码子分析 统计核酸序列中同义密码子使用频率并作图
cai 密码子分析 计算密码子适应指数
chips 密码子分析 统计有效密码子数
palindrome 重复片段查找 寻找核酸序列中反向重复片段
einverted 重复片段查找 寻找核酸序列中反向重复片段
etandem 重复片段查找 寻找核酸序列中的串联重复片段
pepstats 一级结构分析 统计蛋白质序列中不同种氨基酸出现频率
pepinfo 一级结构分析 用图形方式显示蛋白质序列不同氨基酸分布特征
antigenic 一级结构分析 预测蛋白质序列中抗原决定簇
sigcleave 一级结构分析 寻找蛋白质序列中信号肽切割位点
digest 一级结构分析 寻找蛋白质序列中蛋白酶酶切位点
fuzzprot 一级结构分析 寻找蛋白质序列中序列模体
Tmap 二级结构分析 预测蛋白质序列中的跨膜螺旋
topo 二级结构分析 显示跨膜螺旋拓扑结构
pepwheel 二级结构分析 绘制alpha螺旋轮
pepcoil 二级结构分析 预测无规卷曲区域
helixturnhelix 二级结构分析 螺旋-转角-螺旋序列模体分析
garnier 二级结构分析 蛋白质序列二级结构预测
表 8 EMBOSS软件包常用程序Table 8 List of commonly used programs in EMBOSS
EMBOSS网站列出了所有程序的名称和用途,也可用wossname命令按功能分类或字母表顺序列出所有程序。

按功能分类列出所有程序

按字母表顺序列出所有程序
除了EMBOSS开发团队自行编写的程序外,EMBOSS还整合了不少其它常用生物信息软件包,如基于隐马尔可夫模型的蛋白质结构域序列谱构建和结构域识别软件包HEMMER、系统发育分析软件包Phylip及RNA二级结构分析和预测软件包VIENNA等。限于篇幅,本文未介绍这些软件包中程序的用法。
6.3 回顾和展望EMBOSS项目始于上世纪九十年代,初始宗旨是取代序列分析商业软件包GCG。上世纪八十年代,美国威斯康辛大学遗传计算团队(Genetic Computing Group, GCG)开发了基于UNIX的序列分析软件并商业化。该软件包整合了序列比对、酶切位点分析等许多工具,在美国和欧洲等西方国家十分流行[9]。早期的GCG软件包源代码公开,用户可以修改和整合自己的程序。九十年代末,GCG软件包不再公开源代码。为避免GCG商业软件的限制,欧洲分子生物学网络组织EMBnet启动了EMBOSS项目,并很快取代了GCG。有关EMBOSS项目启动和实施过程,以及EMBnet的详细情况,请参阅本刊拟于年内发表的“EMBOSS和EMBnet”一文。
本世纪初,Peter Rice领导的EMBOSS研发团队受聘于欧洲生物信息学研究所,完成了该软件包的主要开发。2009年,EMBOSS项目曾得到英国生物技术和生命科学研究委员会(Biotechnology and Biological Science Research Council, BBSRC)资助,继续进行开发。目前,该软件包开发项目已经结束,由英国transSMART基金会Peter Rice负责维护。显然,作为开源软件,EMBOSS的进一步开发,需要得到生物信息软件开发人员和广大用户的支持。对该软件包开发感兴趣的读者可与Peter Rice直接联系,联系方式请参阅补充材料1。
致谢感谢杨德昌安装和调试EMBOSS软件包,颜林林改正点阵图程序中的错误。感谢樊丽编写的EMBOSS顺口溜。金录佳、李宏博和赵坤认真阅读并校正了初稿中多处文字错误。感谢匿名审稿人宝贵的修改意见。感谢中国科学院北京基因组研究所(国家生物信息中心)对EMBOSS网络教程提供的支持。感谢北京大学生命科学学院、中国农业科学院研究生院和中国科学院大学生命科学学院多年来对“实用生物信息技术”课程的支持。
参考文献
[1] RICE P, LONGDEN I, BLEASBY A. EMBOSS: The European Molecular Biology Open Software Suite[J]. Trends in Genetics, 2000, 16(6): 276-277. DOI:10.1016/s0168-9525(00)02024-2 (
 0)
0)[2] OLSON S A. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite[J]. Briefings in Bioinformatics, 2002, 3(1): 87-91. DOI:10.1093/bib/3.1.87 (
0)[3] MULLAN L J, BLEASBY A J. Short EMBOSS user guide. European Molecular Biology Open Software Suite[J]. Briefings in Bioinformatics, 2002, 3(1): 92-94. DOI:10.1093/bib/3.1.92 (
0)[4] 罗静初. 实用生物信息技术课程教学实例[J]. 生物技术通报, 2015, 31(11): 102-111.
LUO Jingchu. Teaching examples of applied bioinformatics course[J]. Biotechnology Bulletin, 2015, 31(11): 102-111. DOI:10.13560/j.cnki.biotech.bull.1985.2015.07.001 (
0)[5] 罗静初. UniProt蛋白质数据库简介[J]. 生物信息学, 2019, 17(3): 131-144.
LUO Jingchu. A brief introduction to UniProt[J]. Chinese Journal of Bioinformatics, 2019, 17(3): 131-144. DOI:10.12113/j.issn.1672-5565.201903005 (
0)[6] CARVER T, BLEASBY A. The design of Jemboss: A graphical user interface to EMBOSS[J]. Bioinformatics, 2003, 19(14): 1837-1843. DOI:10.1093/bioinformatics/btg251 (
0)[7] ZHU Y, ZHANG Y, LUO J, et al. PPF-1, a post-floral-specific gene expressed in short-day-grown G2 pea, may be important for its never-senescing phenotype[J]. Gene, 1998, 208(1): 1-6. DOI:10.1016/s0378-1119(97)00613-6 (
0)[8] HAMMARSTROM S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues[J]. Seminars in Cancer Biology, 1999, 9(2): 67-81. DOI:10.1006/scbi.1998.0119 (
0)[9] WOMBLE D D. GCG: The Wisconsin Package of sequence analysis programs[J]. Methods in Molecular Biology, 2000, 132: 3-22. DOI:10.1385/1-59259-192-2:3 (
0)