 , 陈云雨1
, 陈云雨1
1. 皖南医学院 药物筛选与评价研究所, 安徽 芜湖 241002;
2. 吉林大学中日联谊医院 风湿免疫科, 吉林 长春 130033
收稿日期:2020-03-19;接收日期:2020-05-19;网络出版时间:2020-05-27
基金项目:国家自然科学基金(No. 81703546),安徽省自然科学基金(No. 1808085QH265),吉林省科技发展计划项目(No. 20160520045JH),安徽省高校自然科学研究重大项目(No. KJ2019ZD30),安徽省重点研究与开发计划项目(No. 202004a07020041)资助
摘要:保罗样激酶1(Polo-like kinase 1, Plk1)与恶性肿瘤的发生与发展密切相关,被认为是肿瘤分子靶向治疗中最具潜力的重要靶标之一。目前,针对Plk1激酶结构域(Kinase domain, KD)设计Plk1抑制剂已成为研究热点,其中部分小分子抑制剂已进入Ⅰ/Ⅱ期临床研究并展现出良好的抗癌前景。尽管Plk1 KD结构域抑制剂具有一定的靶标选择性,但鉴于作为ATP结合口袋的KD结构域在众多激酶结构中的高度保守性和易导致交叉耐药等问题,这使开发高选择性的Plk1 KD结构域抑制剂面临极大的挑战。保罗盒结构域(Polo-Box domain, PBD)作为Plk1特有的底物结合域,在调控Plk1的亚细胞定位中具有重要功能,被认为是未来高选择性Plk1抑制剂开发的理想靶标。文中对Plk1 PBD的分子结构、生物学功能和相关抑制剂的研究进展进行了综述和展望,以期为靶向Plk1 PBD结构域抑制剂的分子设计提供有益的借鉴和参考。
关键词:保罗样激酶1抑制剂保罗盒结构域蛋白质-蛋白质相互作用变构调节剂分子靶向治疗
Mining Polo-Box domain of Polo-like kinase 1 as a new therapeutic target for cancer
Zhenghao Fu1, Meihua Su2, Xiaoping Liu1
, Yunyu Chen1 1. Institute for Drug Screening and Evaluation, Wannan Medical College, Wuhu 241002, Anhui, China;
2. Department of Rheumatology, China-Japan Union Hospital of Jilin University, Changchun 130033, Jilin, China
Received: March 19, 2020; Accepted: May 19, 2020; Published: May 27, 2020
Supported by: National Natural Science Foundation of China (No. 81703546), Natural Science Foundation of Anhui Province (No. 1808085QH265), Jilin Scientific and Technological Development Program (No. 20160520045JH), University Natural Science Research Project of Anhui Province (No. KJ2019ZD30), Key Technologies Research and Development Program of Anhui Province (No. 202004a07020041)
Author: Xiaoping Liu. Tel: + 86-553-3932601; E-mail: liuxiaoping@wnmc.edu.cn.
Corresponding author: Xiaoping Liu. Tel: + 86-553-3932601; E-mail: liuxiaoping@wnmc.edu.cn.
Abstract: Polo-like kinase 1 (Plk1) is widely regarded as one of the most promising targets for cancer therapy due to its essential role in cell division and tumor cell survival. At present, most Plk1 inhibitors have been developed based on kinase domain, some of which are in clinical trial. However, inhibitors targeting kinase domain face off-target effect and drug resistance owing to the conserved nature and the frequent mutations in the ATP-binding pocket. In addition to a highly conserved kinase domain, Plk1 also contains a unique Polo-Box domain (PBD), which is essential for Plk1's subcellular localization and mitotic functions. Inhibitors targeting Plk1 PBD show stronger selectivity and less drug resistance for cancer therapy. Therefore, Plk1 PBD is an attractive target for the development of anti-cancer agents. In this review, we will summarize the up-to date drug discovery for targeting Plk1 PBD, including the molecular structure and cellular functions of Plk1 PBD. Small-molecule inhibitors targeting Plk1 PBD not only provide an opportunity to specifically inhibit Plk1 activity for cancer treatment, but also unveil novel biological basis regarding the molecular recognition of Plk1 and its substrates.
Keywords: Polo-like kinase 1 inhibitorPolo-Box domainprotein-protein interactionsallosteric agentmolecular targeted therapy
Polo-like kinases (Plks) are a conserved family of serine/threonine kinases regulating multiple essential steps in mitosis[1]. Five mammalian Plk family members, Plk1-5, have been identified so far. Among them, Plk1-4 are closely related, which are characterized by a unique C-terminal noncatalytic region containing one (Plk4) or two (Plk1-3) tandem Polo boxes and a highly conserved N-terminal catalytic kinase domain (KD, also named by ATP-binding pocket)[2-3]. The Polo-Box domain (PBD) of Plk1 (PBD1) interacts with phosphorylated substrates containing a consensus S-pS/pT-P/X motif[4-5], which has been shown to be crucial for its subcellular localization and mitotic functions. In contrast to Plk1-4 family members, Plk5 is a PBD-containing protein that lacks the kinase domain (Fig. 1).
 |
| Fig. 1 Polo-like kinases in human cells. Schematic representation of the five identified Polo-like kinases (Plks) in human cells (Plk1-5). The protein structure of each Plk is aligned using kinase domain (green) and Polo-Box domain (yellow). The key residues of kinase domain and Polo-Box domain for substrates recognition are shown in figure. |
| 图选项 |
Plk1 regulates several critical mitotic events, such as mitotic entry, centrosome maturation, spindle assembly, chromosome segregation and cytokinesis [1, 6-7]. Overexpression of Plk1 has been reported in many human cancers, which provides a possible basis for selective elimination of cancer cells. Therefore, Plk1 is generally regarded as an anti-cancer drug target as well as a potential prognostic biomarker for cancer patients[8-10]. Inhibition of Plk1 activity causes mitotic arrest and apoptotic cell death in most cancer cell lines. Plk1 inhibitors reduce tumor proliferation, and have been shown little effect on normal cells using mouse xenograft models[11]. Therefore, Plk1 is a very attractive therapeutic target for the discovery and development of small-molecule anti-cancer drugs.
In contrast to Plk1, Plk2 and Plk3 have been identified as tumor suppressors to prevent mitotic catastrophe and preserve genomic integrity[7-12]. Moreover, significantly lower expression of Plk2 has been observed in a wide range of B-cell neoplasms due to CpG methylation-dependent transcriptional silencing of Plk2, supporting the role of Plk2 as a tumor suppressor in hematopoietic diseases[13]. The function of Plk4 is not fully understood yet, but evidence suggests that it may ensure genome stability through the regulation of centriole duplication[14-15]. Plk5 is highly expressed in the central nervous system and has been reported as a glioblastoma suppresser[16]. Therefore, the highly selective inhibition of Plk1 activity, but not other Plks, is a perfect strategy for cancer therapy.
Many small-molecule Plk1 inhibitors targeting the kinase domain have been identified and some are being tested in clinical trials for cancer treatment, such as BI6727 (Volasertib)[17-20], ON01910 (Rigosertib)[21-23], BI2536[24-26], GSK461364[27] and HMN-214[28] (Table 1). Recently, volasertib was approved by FDA due to its significant benefit to the treatment of acute myeloid leukemia in combination with cytarabine. Moreover, a phase Ⅱ trial for volasertib in advanced non-small cell lung cancer (NCT00824408) is underway[29-30]. Rigosertib is now in phase Ⅲ trials for second-line treatment of high-risk myelodysplastic syndrome (NCT01928537 and NCT00906334) and phase Ⅱ trials for first-line treatment of low-risk myelodysplastic syndrome (NCT01584531 and NCT01904682). A phase Ⅲ study of rigosertib in combination with gemcitabine is also underway in patients with metastatic pancreatic cancer (NCT01360853)[30]. The ATP-binding pocket of protein kinases is a classical target for kinase inhibitors development and has been validated by several successful drug discovery programs. However, these inhibitors usually face drug resistance due to the frequent mutations in the ATP-binding pocket[29, 31]. Another disadvantage of these inhibitors is the serious off-target effect due to the conserved nature of ATP-binding pocket of all kinases. Therefore, the development of effective and Plk1-selective inhibitors remains challenging.
Table 1 Ongoing Plk1 small-molecule inhibitors in clinical trials
| Compound | Mode of action | Phase | Clinical indications | References |
Volasertib (BI6727) 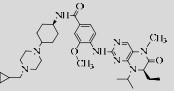 | ATP-competitive inhibitor | Phase Ⅲ | Acute myeloid leukemia | [17-20] |
Rigosertib (ON01910)  | Affects microtubule dynamics | Phase Ⅲ | Advanced solid tumors, myelodysplastic syndrome | [21-23] |
BI2536  | ATP-competitive inhibitor | PhaseⅡ | Non-small cell lung cancer, acute myeloid leukaemia | [24-26] |
GSK461364  | ATP-competitive inhibitor | PhaseⅠ | Advanced solid tumors | [27] |
HMN-214  | A prodrug that alters spatial distribution of Plk1 (no direct catalytic inhibition) | PhaseⅠ | Advanced solid tumors | [28] |
表选项
In addition to a highly conserved kinase domain, Plk1 also contains a unique Polo-Box domain, which provides a possible drug discovery target for selectively inhibiting Plk1. Currently, there is an increasing interest in the development of small-molecule inhibitors targeting PBD1 for treating cancer diseases. Herein, we will review the up-to date progress in this field.
1 Plk1 PBD and drug discoveryThe crystal structure of human PBD1 shows that it contains two β6α motifs that comprise two Polo-Box regions (PB1 and PB2). In spite of the fact that the amino acid sequences of the two Polo boxes exhibit only 20%-25% similarity, the structures of the two motifs are quite similar. The PBD1 also contained a Polo cap region consisting of α-helix connected via a linker (L1) to the first β-strand of PB1 and a second linker (L2) connecting the two Polo-box repeats, helping to hold both Polo-boxes in the correct orientation. The crystal structures of the PBD1 also reveal that the phosphopeptide (Poloboxtide, see 3.1 section) binding site is located at one end of a shallow cleft between the two β-sheets only within the highly conserved region on the PBD1 surface (Fig. 2A). The residues that contact the phosphate group directly are His538 and Lys540 from PB2, whose side chains form pincer-like hydrogen bonds that chelate the phosphate oxygens (Fig. 2B)[5]. Additionally, the kinase domain and PBD1 are mutually inhibited in vivo and binding of the PBD1 to substrates with primed phosphorylation sites simultaneously activates the kinase domain to enhance the biological activities of Plk1 (Fig. 2C). The conformational change of L2 serves as the key switch between the on and off states of Plk1[32].
 |
| Fig. 2 Molecular structure and cellular functions of Plk1 PBD (PBD1). (A) The crystal structure of the PBD1 is shown in a ribbons representation in complex with Poloboxtide (shown in yellow). Two Polo boxes (PB1 and PB2) are colored in red and purple, respectively, and the Polo-cap region that accommodates Poloboxtide is colored in grey. (B) Schematic representation of pincer-like hydrogen bonds (dotted lines) between the phosphate and His538/Lys540 of PBD1. Hydrogen bond lengths are given in angstroms. (C) A model for Plk1 regulation by the PBD1. PB1 and PB2 are colored by orange, kinase domain and a representative phosphopeptide are colored by blue and purple with phosphate in red, respectively. Inhibitory interactions between the PBD1 and the kinase domain in the basal state (left) are relieved by phosphopeptide binding (right). Reproduced with permission[5]. |
| 图选项 |
Moreover, Plk1 is auto-inhibited through KD/PBD binding in order to regulate Plk1 functions during mitosis. When the conserved threonine residue (Thr210) in T-loop of the Plk1 kinase domain is phosphorylated by Aurora A, the PBD1 will be released from auto-inhibition along with recruitment of phospho-substrate, allowing the initiation of Plk1 activation (Fig. 2C)[5]. Therefore, the PBD1 is crucial for substrate interaction and through its PBD1, Plk1 associates with a large number of subcellular proteins, such as the centrosomes, kinetochores, spindle apparatus and Golgi, which in turn regulates the kinase activity of Plk1[33]. Unlike the highly conserved KD in Plk1, PBD1 is a unique domain, which provides a much more compelling site to selectively inhibit the localization and biological functions of Plk1. The significant role of PBD1 in Plk1 subcellular localization and functional regulation renders the drug development superiority by targeting this domain. Indeed, several small-molecule inhibitors and peptide mimics targeting PBD1 have been reported, and Table 2 briefly summarizes all emerging small-molecule PBD1 inhibitors so far.
Table 2 Emerging small-molecule Plk1 inhibitors targeting Polo-Box domain
| Compound | IC50 values against PBDs | Key binding residues | References |
Poloxin 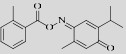 | 4.8±1.3 μmol/L (PBD1) 18.7±1.8 μmol/L (PBD2) 53.9±8.5 μmol/L (PBD3) | Val411, Asn533 and Lys540 | [36-38] |
Thymoquinone (TQ)  | 1.14±0.04 μmol/L (PBD1) 1.90±0.1 μmol/L (PBD2) 22.4±0.8 μmol/L (PBD3) | Val411, Asn533 and Lys540 | [36, 38] |
Poloxipan  | 3.2±0.3 μmol/L (PBD1) Pan-inhibitor for PBD2 and PBD3 | Not determined (N.D.) | [39] |
Purpurogallin (PPG)  | 0.3 μmol/L (PBD1) Most efficiently exhibits activity against PBD2, but weak activity against PBD3 | Trp411, His538 and Lys540 | [40, 44] |
(-)-epigallocatechin (EGC)  | 10 μmol/L (PBD1) Most efficiently blocks PBD2 with less inhibition for PBD3 | Leu491, His538 and Lys540 | [45] |
(-)-epigallocatechin gallate (EGCG)  | N.D. | Leu491, His538 and Lys540 | [45] |
(+)-gallocatechin (GA)  | N.D. | Leu491, His538 and Lys540 | [45] |
(+)-catechin (CAT)  | N.D. | Leu491, His538 and Lys540 | [45] |
T521  | (1.22±0.13) μmol/L (PBD1) > 500 μmol/L (PBD2 and PBD3) | Covalent binding (Lys209, Lys388, and Lys574) | [46] |
表选项
More interestingly, the overall structure of the Plk2 PBD (PBD2) is highly similar to that of the PBD1, which is composed by two Polo boxes each contain β6α structures that form a 12-stranded β sandwich domain. The edge of the interface between the two Polo boxes forms the phosphorylated Ser-pSer/pThr motifs binding cleft. On the other hand, only the peripheral regions around the core binding cleft of the PBD2 is distinct from that of the PBD1, which might confer the substrate specificity of the PBDs in Polo-like kinase family (Fig. 3). Importantly, two residues including Lys607 and Tyr626 involved in peripheral regions are highlighting for this substrate recognition of PBD2[34-35]. To date, the crystal structure of Plk3 PBD (PBD3) has not been reported yet.
 |
| Fig. 3 Comparison of the peripheral regions of the core phosphopeptide binding cleft in PBD1 (green) and PBD2 (yellow). The core region is labeled by red solid line ellipse, and the peripheral region is labeled by red dash line ellipse. Lys607 and Tyr626 in the peripheral region of PBD2 are highlighted. Reproduction with permission[34]. |
| 图选项 |
2 Plk1 PBD small-molecule inhibitors2.1 Thymoquinone (TQ) and its derivative, PoloxinReindl et al. have developed a fluorescence polarization (FP) assay based on the binding of fluorophore-labeled peptide comprising of an optimal recognition motif to PBD1[36]. Screening of diverse chemical libraries for compounds, which could interfere with the PBD1 binding, led to the discovery of TQ and its derivative Poloxin. These are the first reported small-molecule inhibitors that block Plk1 PBD-mediated protein-protein interactions. Poloxin inhibits the PBD1 in vitro using FP assay (IC50 of 4.8 μmol/L), as well as that of Plk2 and Plk3 to a slightly less extent (IC50 of 18.7 and 53.9 μmol/L, respectively), but Poloxin does not appear to significantly inhibit other types of phosphopeptide- binding domains such as Chk2 FHA, Pin1 WW, and STAT3 SH2 domains. Both compounds cause Plk1 mislocalization, chromosome congression defects, mitotic arrest, and apoptosis in HeLa cells[36]. Moreover, Poloxin induces centrosome fragmentation and abnormal spindle and chromosome misalignment, which induces mitotic arrest, followed by apoptosis, and significantly suppresses tumor growth in xenograft mouse models[37].
Recently, Yin et al. reported TQ/PBD1 crystal structure and revealed the molecular basis of TQ/PBD1 recognition[38]. The crystallographic studies show that the phosphoserine/phosphothreonine recognition site of the Polo-Box domain is the binding pocket for TQ, which consists of a hydrophobic half (Val411, Trp414, Leu490, and Leu491) and a positively charged half (His538, Lys540, and Arg557) where the Lys540-His538 pincer clinches phosphopeptides by the phosphate. TQ displaces phosphopeptides bound with the Polo-Box domain in a slow but noncovalent binding mode. A conserved water bridge and a cation-π interaction have been found as the competition strategy against the phosphate group (Fig. 4). The binding of TQ is in effect a displacement of several crystallographic waters. Together, the complex crystal structure offers an insight into the binding mode of TQ competing with phosphopeptide interacting with PBD1. The phospho-mimetic mechanism may be applicable to the general inhibitor design for drugs targeting PBD1 and other phospho-binding domains.
 |
| Fig. 4 The crystal structure of TQ/PBD1 complex. (A) Schematic representation of hydrogen bonds between TQ (yellow) and a conserved structural water (W1). Atoms are colored by model except noncarbon atoms of the PBD1/TQ side chains (N, blue; O, red). Hydrogen bonds are given as red dashed lines. Arrows show the shift of Leu491. (B) Shortest intermolecular distances between Lys540+ and TQ are represented as dashed lines. Reproduced with permission[38]. |
| 图选项 |
2.2 PoloxipanPoloxipan, a pan-specific inhibitor of the PBDs of Plk1-3, was recently screened from diverse chemical libraries against the PBD1[39]. Poloxipan inhibits the functions of PBDs with the IC50 values of 3.2 μmol/L for PBD1, 1.7 μmol/L for PBD2, and 3.0 μmol/L for PBD3 in FP assay, and exhibits a significant less extent for other peptide motifs (i.e. Chk2 FHA, Pin1 WW, and STAT1, 3, 5 SH2). Unfortunately, the use of higher concentrations of Poloxipan was not feasible due to its limited solubility in aqueous buffers. The cellular effects of Poloxipan are very similar to that of TQ and Poloxin, especially the mitotic arrest, chromosome alignment defect, Plk1 subcellular localization and centrosome fragmentation. This observation further supports that functional inhibition of PBD1 by small molecules is an effective strategy for future cancer treatment.
2.3 Purpurogallin (PPG)Watanabe et al. have developed an enzyme linked immunosorbent assay (ELISA) based on the binding of the Venus-PBD1 to the phosphopeptide containing PBD1 consensus motif[40]. The Wee1A phosphopeptide could covalently bind to maleimide-activated 96-well plates prior to screening. The bacterial lysates expressing Venus-PBD1 were mixed with the test compounds and then loaded into each well; unbound PBD1 was washed away, and bound PBD1 was quantitated by spectrofluorometry (excitation, 485 nm; emission, 530 nm). When small molecules that compete with the phosphopeptide for PBD1 binding are present in the assay, Venus-PBD1 will exhibit less fluorescent signal. Purpurogallin (PPG) was identified as a potent PBD1 inhibitor through this screening assay from 2 500 natural products. PPG most efficiently exhibits activity against PBD1 and PBD2 (IC50=0.3 μmol/L), but weak activity against PBD3 using this developed ELISA-like assay. PPG not only delays the onset of mitosis but also prolongs the progression of mitosis in HeLa cells. Although apparently normal bipolar spindles were formed even in the presence of PPG, the perturbation of chromosome alignment at metaphase plates activated the spindle assembly checkpoint pathway. These results demonstrate the predominant role of PBD1-dependent binding on timely chromosome congression at metaphase and PPG may serve as a novel potent anti-cancer agent. Interestingly, PPG-like compounds lacking the 2-hydroxyl group failed to exhibit the inhibitory activity, indicating that the 2-hydroxyl group of PPG is essential for the observed activity. However, whether PPG could efficiently suppress the tumor growth in an in vivo mouse model by targeting PBD1 has not been explored, PPG may become an attractive lead compound of Plk1 inhibition. Notably, current studies showed that PPG could strongly suppress esophageal squamous cell carcinoma (ESCC) growth by directly targeting the mitogen-activated protein kinase 1/2 (MEK1/2) signaling pathway in vitro and in vivo, supporting a general idea of multi-targets in human diseases therapeutic for PPG, a natural compound extracted from nutgalls and oak bark[41-43].
Docking was used to explore the binding modes of PPG[44]. The results suggest that the negatively charged enolized hydroxyl group of the bound PPG forms an electrostatic interaction with the positively charged His538. The other three phenolic hydroxyl groups and the carbonyl group form four hydrogen bonds with the positively charged Lys540, the backbone NH of Trp414, the indole ring of Trp414, and the backbone carbonyl of Leu491. Moreover, the phenyl ring and one double bond in the tropolone ring of PPG form π-π stacking interactions with the pyrrole ring and the phenyl ring of Trp414, respectively (Fig. 5). The analogue of PPG lacking the 2-hydroxyl group was unable to form reasonable docking poses, underlying the importance of the 2-hydroxyl group for PBD1 inhibition. In order to get a deeper insight into the PPG-PBD1 interaction, the crystal structure is urgently required.
 |
| Fig. 5 The binding mode of PPG to the PBD1 generated by Schrodinger's induced fitting docking protocol. Hydrogen bonds are shown as red dashed lines. Reproduced with permission[44]. |
| 图选项 |
2.4 Green tea catechinsBy PBD1 structure-based in silico virtual screening, (-)-epigallocatechin (EGC), one of the main components of green tea polyphenols, was identified as a potent PBD1 inhibitor with IC50 value of 10 μmol/L in FP assay[45].
To understand the interaction between EGC and the PBD1, Shan et al. also tested the effects of the other major green tea catechins including (-)-epigallocatechin gallate (EGCG), (+)-gallocatechin (GA), and (+)-catechin (CAT)[45]. Compared with EGC, EGCG has higher inhibitory activity, suggesting that the additional gallate group contributes to the binding with the PBD1. The lower inhibitory activity for GA indicates that the configuration of 3-hydroxyl group plays a role in the binding of catechins with the PBD. More strikingly, CAT has much lower inhibitory activity compared with GA, which indicates that the hydroxyl groups on B ring are essential for the inhibition of PBD-dependent recognition.
2.5 T521By FP screening assay, T521 was identified as a specific small-molecule inhibitor of PBD1 from the diverse chemical libraries of 20 000 compounds in Prof. Si's laboratory[46]. Using an in vivo PBD substrate Map205, T521 could efficiently block the interaction between PBD1 and GST-Map205PBM in vitro. HeLa cells treated with T521 exhibited dramatic mitotic defects, including Plk1 mislocalization, centrosome fragmentation, chromosome misalignment, and mitotic arrest, which are the consequences of inhibition of Plk1 by targeting PBD1. Importantly, T521 suppresses the growth of A549 cells in xenograft nude mice. Unlike other known PBD1 inhibitors, T521 specially inhibits the PBD1, shows much less effect on the PBD2 and PBD3, indicating its specificity toward Plk1.
Interestingly, the T521 inhibition of PBD1 was time and temperature dependent, which implies the possibility of covalent binding mode. HPLC-Q-TOF mass spectrometry analysis was performed and the intact mass of Plk1 protein increased by 907.05 Da, suggesting covalent addition of three T521 fragments (301 Da) to Plk1, and covalent binding sites of Plk1 are Lys209, Lys388 and Lys574. However, Lys209 and Lys388 binding sites do not locate within the core phosphopeptide binding pocket. In contrast, no addition of T521 to Plk2 or Plk3 was detected after their incubation with T521. The circular dichroism (CD) spectra of PBD1 show the significant secondary structure changes after T521 binding, especially the composition of α-helix. Because the native conformational structure of PBD1 is necessary for phosphopeptide binding, these secondary structure changes caused by T521 covalent binding may contribute to its strong inhibition of PBD1 functions. However, the specificity addition mechanism of covalent binding in T521 remains unclear. Maybe the allosteric regulation strategy in classic or nonclassic binding pocket is a new avenue for the development of next generation PBD1 inhibitors.
3 Plk1 PBD peptide derived inhibitors3.1 Poloboxtide: MQSpTPLPlks PBD from various species including yeast, xenopus, and human exhibit strong affinity to the core sequence S-pS/pT-P/X (X, any amino acid residue)[4-5]. Crystal structure of the PBD1 with bound Poloboxtide, MQSpTPL, shows that the phosphopeptide sits in a pocket formed by PB1 and PB2. Lys540 and His538 residues are the only residues that directly contact with the phosphate group in a form of "pincer grip", which is stabilized by a network of van der Waals interactions and hydrogen bonds between water molecules and other conserved residues within the PBD1. The pThr or pSer containing phosphopeptide interacts in an extended conformation with one end of a planar gap that is constituted between the two PBs. The explanation for the Ser preference at the -1 position seems to be that the side-chain of Ser forms a hydrogen bond with Trp414, a highly conserved residue in all PBDs. Significantly, this finding is consistent with an earlier observation that a Trp414Phe mutation disrupts Plk1 localization to spindle poles and abrogates its functions, further supporting the key role of Trp414 in its ligand binding[47].
3.2 PLHSpTAlthough the fact that S-pS/pT-P/X phosphopeptides optimized for PBD1 binding reveals the key binding residues, the molecular basis for PBD1 binding specificity remains elusive. Among various PBD1-binding proteins, PBD1 binds to the Thr78 region of PBIP1 in a phosphorylation-dependent manner. Systematic analyses of the PBD1-PBIP1 p-T78 motif interaction led to the identification of a minimal PBD1-binding sequence, PLHSpT[48]. Structural analyses of the PBD1 in complex with the minimal p-T78 peptide revealed that the N-terminal Pro residue plays an important role in conferring the specificity by docking its side-chain into a hydrophobic core surrounded by the Trp414, Phe535, and Arg516 residues, while concomitantly participating in a hydrogen bonding interaction between its carbonyl oxygen and the guanidinium moiety of Arg516 of the PBD1.
The PBD2 and PBD3 possess Lys and Tyr residues at positions corresponding to the Arg516 and Phe535 residues in PBD1, respectively, failing to interact with the N-terminal Pro residue. PLHSpT exhibits a dose-dependent PBD1 inhibition with a dissociation constant (Kd) of 0.44 μmol/L[48]. However, the hydrolytic lability of phosphoryl esters to phosphatases limits the use of phosphopeptides in cellular contexts. Development of hydrolytically- stable mimetic peptides, in which the labile phosphoryl ester oxygen is replaced with non- hydrolysable methylene ordifluoromethylene groups, offers a way to circumvent this limitation. The phosphatase-resistant pThr mimetic (2S, 3R)-2- amino-3-methyl-4-phosphonobutyric acid (Pmab)- containing peptide, PLHS-Pmab, binds to PBD1 with an undiminished affinity and specificity, and efficiently induces mitotic arrest, when microinjected into HeLa cells.
This finding provides the proof of principle that highly selective inhibition of PBD1 can be achievable by small PBD1-binding mimetic peptides and a new avenue for the development of anti-PBD1 therapeutic agents.
3.3 PeptoidBased on the core significance of Pro residence for PLHSpT affinity and specificity, Liu et al. replaced the amino terminal Pro residue of the Plk1 polobox-domain-binding pentapeptide (PLHSpT) with a library of N-alkyl-Gly "peptoids", and identified long-chain tethered phenyl moieties that give greater than two-orders-of-magnitude affinity enhancement[49]. Further simplification by replacing the peptoid residue with appropriate amides gave low-nanomolar affinity N-acylated tetrapeptides.
In an effort to develop improved binding antagonists of the PBD1, Liu et al. further optimized binding affinity of 5-mer peptide PLHSpT using oxime-based post solid-phase peptide diversification of the N-terminal Pro residue[50]. These results show that trans-(4R) phenylbutyloxy substituent on the P1 pyrrolidine ring increases the binding affinity and replacing the oxime functionality is deleterious for high binding affinity.
To improve the pharmacological properties of PLHSpT, Liu et al. replaced, the hydrolytically labile pT residue with the phosphatase-stable pT mimetic, (2S, 3R)-2-amino-3-methyl-4-phosphonobutanoic acid (Pmab) without loss of PBD1 binding affinity[51]. The affinities of n-alkylphenyl byproducts with PBD1 increases roughly with lengthening of the alkyl chain (with the exception of n=6 and n=7) and reached a maximum for n=8. Chain extension beyond this length resulted in a reduction in binding affinity. Further, they solved the cocrystal structure of PBD1 in complex with the modified peptide 4j (Fig. 6A), and showed that the most substantial movement occurred in the orientation of the Tyr417 and Tyr481 aryl ring, and this movement had profound effects on the topology of the protein surface, resulting in the revelation of a new binding channel that had previously been occluded (Fig. 6B). The availability of this hydrophobic channel was not anticipated from previously reported crystal structure. Most importantly, these results highly consist with a previously identified binding pocket using a crystal packing by ?led? P[52]. This flexible binding site consists of seven residues: Val415, Leu478, and Phe482 form the bottom of the pocket whilst Tyr417, Tyr421, Tyr481, and Tyr485 form the sides of the pocket (Fig. 6C). The conformational changes in the side-chain of Tyr417 and Try481 confers a much shallow and plastic cavity to anchor substrates. Maybe the role of this binding site has a synergistic action when the core binding pocket works. This newly indentified binding pocket may provide a new paradigm for the discovery of novel PBD1 inhibitors in the future.
 |
| Fig. 6 A new binding site identified by a co-crystal structure of PBD1/peptide 4j complex. (A) The chemical structure modified peptide 4j. (B) The key residues involved in the structure of PBD1/peptide 4j complex and the displacement of Tyr417 and Tyr481 phenyl groups are rendered. Reproduction with permission[51]. (C) Seven key residues forming a new binding site in different conformations observed in the crystal structures and the mobile residues are shown in yellow. Reproduction with permission[52]. |
| 图选项 |
Moreover, N-terminal PEGylated version of this peptide containing a hydrolytically stable phosphothreonyl residue (pT) bound the PBD1 with affinity equal to that of the non-PEGylated parent, but showed markedly less interaction with the PBD2 and PBD3. Compared to non-PEGylated version, N-terminal PEGlated peptide indeed enhanced its pharmaceutical properties. However, all these reported peptides strongly inhibited the functions of Plk1 by targeting PBD1 in vitro, massive efforts will be needed to further improve its druggability in vivo, especially the experiments in animal models for the coming preclinical development.
4 Summary and future outlookCancer cells display characteristics of sustaining proliferative signaling pathway, evading growth suppressors, and resisting cell death[53], and Plk1 is involved in all these processes during cancer cell survivals. Accumulating evidence suggests that Plk1 is an appealing therapeutic target for cancers, and a number of small-molecule inhibitors that target the kinase domain of Plk1 are currently under preclinical or clinical trials[7]. However, ATP-competitive Plk1 inhibitors show poor selectivity, owing to the well conserved kinase domain of Plks and other kinases. In addition, drug resistance is commonly associated with these Plk1 inhibitors targeting the kinase domain[11]. The PBD is unique to Plks family and it has been shown to regulate Plk1's subcellular localization for its mitotic functions. Therefore, the PBD1 represents an attractive target for the development of Plk1-specific inhibitors.
So far, most emerging PBD1 inhibitors are natural products with limited chemical structure diversities. Systematic analysis of structure-activity relationship is urgently needed to develop more selective and effective PBD1 inhibitors. The binding mode represented by TQ/Poloxin is likely a general binding mechanism of PBD1 inhibitors. Therefore, the phosphoserine/threonine recognition site of PBD1 is a powerful binding pocket for small-molecule PBD1 inhibitors development. As noted previously, protein-protein interactions are notoriously difficult to target with small molecules as large, discontinuous surfaces are often involved[52, 54]. Furthermore, the overall structure of the PBD2 is highly similar to that of the PBD1 except for the peripheral regions, so these reported small molecule inhibitors targeting PBD1 exhibited the modest selectivity toward PBD2 and PBD3 in vitro. Considering the cellular functions of Plks and the structure similarities, how to improve the selectivity of PBD1 inhibitors remains challenging. In the near future, more attentions should focus on the unique regions of PBD1, such as the peripheral regions or the currently identified new binding site in Fig. 5. Seeking more efficient PBD1 inhibitors via chemical modifications in lead compounds or high throughput screening (HTS) strategy will be still urgently encouraged. Moreover, the allosteric agents including the specific covalent inhibitors could result in a structure disaster to selectively impede the functions of PBD1, so the allosteric modulators and covalent kinase inhibitors (CKI) also could be regard as a promising strategy for future anti-Plk1 therapy[46, 55-57].
Currently, protein-protein interactions have become promising anti-cancer therapeutic targets[54, 58-59], the identification of PBD1 inhibitors would expand the landscape of new groups of inhibitors targeting PBD1 for cancer therapy. Many peptide inhibitors of PBD1 have been explored and exhibited the promising activities against PBD1 cellular functions in vitro[5, 47-52], but they generally have a shorter half-life, unexpected bioavailability, poor stability and lower membrane permeability for the coming preclinical application[60-61]. Therefore, the urgent efforts will be needed to conquer these highlighting drawbacks and improve the druglike properties of peptide/peptoid inhibitors.
In summary, PBD1 is a potential and promising target for the development of highly selective anti-Plk1 inhibitors for cancer therapy. Better understanding of the binding mode of PBD1 inhibitors may reveal new insights into the development of potent PBD1 inhibitors. Most likely a set of novel PBD1 inhibitors would make a significant contribution to future cancer research and precision therapeutics.
AcknowledgementsWe are sincerely grateful to Dr. Jing Zhang (Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing) and Prof. Yan-chang Wang (Department of Biomedical Sciences, College of Medicine, Florida State University, United States) for their insightful comments and helping readings of the manuscript.
AbbreviationsATP: adenosine triphosphate; Chk2: checkpoint kinase 2; FHA, fork head-associated domain; GST: glutathione-S-transferase; IC50: half maximal inhibitory concentration; Map205: microtubule- associated protein 205; PBM: Polo-Box domain binding motif; PEG, polyethylene glycol; Pin1: peptidyl-prolyl cis/trans isomerase NIMA- interacting 1; SH2: Src homology domain 2; STAT: signal transducer and activator of transcription.
REFERENCES
| [1] | Colicino EG, Hehnly H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton, 2018, 75(11): 481-494. DOI:10.1002/cm.21504 |
| [2] | De Cárcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle, 2011, 10(14): 2255-2262. DOI:10.4161/cc.10.14.16494 |
| [3] | Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene, 2005, 24(2): 248-259. DOI:10.1038/sj.onc.1208280 |
| [4] | Elia AEH, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science, 2003, 299(5610): 1228-1231. DOI:10.1126/science.1079079 |
| [5] | Elia AEH, Rellos P, Haire LF, et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell, 2003, 115(1): 83-95. DOI:10.1016/S0092-8674(03)00725-6 |
| [6] | Tischer J, Gergely F. Anti-mitotic therapies in cancer. J Cell Biol, 2019, 218(1): 10-11. DOI:10.1083/jcb.201808077 |
| [7] | Lee SY, Jang C, Lee KA. Polo-like kinases (Plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev Reprod, 2014, 18(1): 65-71. |
| [8] | Liu ZX, Sun QR, Wang XS. PLK1, A potential target for cancer therapy. Transl Oncol, 2017, 10(1): 22-32. DOI:10.1016/j.tranon.2016.10.003 |
| [9] | Fu Z, Wen DH. The emerging role of Polo-like kinase 1 in epithelial-mesenchymal transition and tumor metastasis. Cancers, 2017, 9(10): 131. |
| [10] | Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer, 2017, 17(2): 93-115. DOI:10.1038/nrc.2016.138 |
| [11] | Park JE, Hymel D, Burke TR Jr, et al. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents[version 1; peer review: 4 approved].. F1000Res, 2017, 6: 1024. DOI:10.12688/f1000research.11398.1 |
| [12] | Yang Y, Bai JX, Shen RL, et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1α under hypoxic conditions. Cancer Res, 2008, 68(11): 4077-4085. DOI:10.1158/0008-5472.CAN-07-6182 |
| [13] | Syed N, Smith P, Sullivan A, et al. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood, 2006, 107(1): 250-256. DOI:10.1182/blood-2005-03-1194 |
| [14] | Yamamoto S, Kitagawa D. Self-organization of Plk4 regulates symmetry breaking in centriole duplication. Nat Commun, 2019, 10: 1810. DOI:10.1038/s41467-019-09847-x |
| [15] | Maniswami RR, Prashanth S, Karanth AV, et al. PLK4: a link between centriole biogenesis and cancer. Expert Opin Ther Targets, 2018, 22(1): 59-73. DOI:10.1080/14728222.2018.1410140 |
| [16] | De Cárcer G, Escobar B, Higuero AM, et al. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol Cell Biol, 2011, 31(6): 1225-1239. DOI:10.1128/MCB.00607-10 |
| [17] | Rudolph D, Steegmaier M, Hoffmann M, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res, 2009, 15(9): 3094-4102. DOI:10.1158/1078-0432.CCR-08-2445 |
| [18] | D?hner H, Lübbert M, Fiedler W, et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood, 2014, 124(9): 1426-1433. DOI:10.1182/blood-2014-03-560557 |
| [19] | Schwermer M, Dreesmann S, Eggert A, et al. Pharmaceutically inhibiting polo-like kinase 1 exerts a broad anti-tumour activity in retinoblastoma cell lines. Clin Exp Ophthalmol, 2017, 45(3): 288-296. |
| [20] | Ottmann OG, Müller-Tidow C, Kr?mer A, et al. Phase Ⅰ dose-escalation trial investigating volasertib as monotherapy or in combination with cytarabine in patients with relapsed/refractory acute myeloid leukaemia. Br J Haematol, 2019, 184(6): 1018-1021. DOI:10.1111/bjh.15204 |
| [21] | Gumireddy K, Reddy MVR, Cosenza SC, et al. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell, 2005, 7(3): 275-286. DOI:10.1016/j.ccr.2005.02.009 |
| [22] | Garcia-Manero G, Fenaux P, Al-Kali A, et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol, 2016, 17(4): 496-508. DOI:10.1016/S1470-2045(16)00009-7 |
| [23] | Navada SC, Fruchtman SM, Odchimar-Reissig R, et al. A phase 1/2 study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk Res, 2018, 64: 10-16. DOI:10.1016/j.leukres.2017.11.006 |
| [24] | Steegmaier M, Hoffmann M, Baum A, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol, 2007, 17(4): 316-322. DOI:10.1016/j.cub.2006.12.037 |
| [25] | Awad MM, Chu QSC, Gandhi L, et al. An open-label, phase Ⅱ study of the polo-like kinase-1 (Plk-1) inhibitor, BI 2536, in patients with relapsed small cell lung cancer (SCLC). Lung Cancer, 2017, 104: 126-130. DOI:10.1016/j.lungcan.2016.12.019 |
| [26] | Ellis PM, Chu QS, Leighl N, et al. A phase Ⅰ open-label dose-escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer. Clin Lung Cancer, 2013, 14(1): 19-27. DOI:10.1016/j.cllc.2012.04.003 |
| [27] | Olmos D, Barker D, Sharma R, et al. Phase Ⅰ study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res, 2011, 17(10): 3420-3430. DOI:10.1158/1078-0432.CCR-10-2946 |
| [28] | Garland LL, Taylor C, Pilkington DL, et al. A phase Ⅰ pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin Cancer Res, 2006, 12(17): 5182-5189. DOI:10.1158/1078-0432.CCR-06-0214 |
| [29] | Strebhardt K, Becker S, Matthess Y. Thoughts on the current assessment of Polo-like kinase inhibitor drug discovery. Expert Opin Drug Discov, 2015, 10(1): 1-8. |
| [30] | Liu XQ. Targeting Polo-like kinases: a promising therapeutic approach for cancer treatment. Transl Oncol, 2015, 8(3): 185-195. DOI:10.1016/j.tranon.2015.03.010 |
| [31] | Park JE, Kim TS, Meng LJ, et al. Putting a bit into the polo-box domain of polo-like kinase 1. J Anal Sci Technol, 2015, 6: 27. DOI:10.1186/s40543-015-0069-y |
| [32] | Xu J, Shen C, Wang T, et al. Structural basis for the inhibition of Polo-like kinase 1. Nat Struct Mol Biol, 2013, 20(9): 1047-1053. DOI:10.1038/nsmb.2623 |
| [33] | Lowery DM, Mohammad DH, Elia AEH, et al. The Polo-box domain: a molecular integrator of mitotic kinase cascades and Polo-like kinase function. Cell Cycle, 2004, 3(2): 128-131. |
| [34] | Shan HM, Wang T, Quan JM. Crystal structure of the polo-box domain of polo-like kinase 2. Biochem Biophys Res Commun, 2015, 456(3): 780-784. DOI:10.1016/j.bbrc.2014.11.125 |
| [35] | Kim JH, Ku B, Lee KS, et al. Structural analysis of the polo-box domain of human Polo-like kinase 2. Proteins, 2015, 83(7): 1201-1208. DOI:10.1002/prot.24804 |
| [36] | Reindl W, Yuan JP, Kr?mer A, et al. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol, 2008, 15(5): 459-466. DOI:10.1016/j.chembiol.2008.03.013 |
| [37] | Yuan JP, Sanhaji M, Kr?mer A, et al. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol, 2011, 179(4): 2091-2099. DOI:10.1016/j.ajpath.2011.06.031 |
| [38] | Yin Z, Song YL, Rehse PH. Thymoquinone blocks pSer/pThr recognition by Plk1 Polo-box domain as a phosphate mimic. ACS Chem Biol, 2013, 8(2): 303-308. DOI:10.1021/cb3004379 |
| [39] | Reindl W, Yuan JP, Kr?mer A, et al. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem, 2009, 10(7): 1145-1148. DOI:10.1002/cbic.200900059 |
| [40] | Watanabe N, Sekine T, Takagi M, et al. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem, 2009, 284(4): 2344-2353. DOI:10.1074/jbc.M805308200 |
| [41] | Xie XM, Zu XY, Liu FF, et al. Purpurogallin is a novel mitogen-activated protein kinase kinase 1/2 inhibitor that suppresses esophageal squamous cell carcinoma growth in vitro and in vivo. Mol Carcinog, 2019, 58(7): 1248-1259. |
| [42] | Glanzer JG, Endres JL, Byrne BM, et al. Identification of inhibitors for single-stranded DNA-binding proteins in eubacteria. J Antimicrob Chemother, 2016, 71(12): 3432-3440. DOI:10.1093/jac/dkw340 |
| [43] | Patel S, Rauf A, Khan H. The relevance of folkloric usage of plant galls as medicines: Finding the scientific rationale. Biomed Pharmacother, 2018, 97: 240-247. DOI:10.1016/j.biopha.2017.10.111 |
| [44] | Liao CZ, Park JE, Bang JK, et al. Exploring potential binding modes of small drug-like molecules to the Polo-Box domain of human Polo-like kinase 1. ACS Med Chem Lett, 2010, 1(3): 110-114. DOI:10.1021/ml100020e |
| [45] | Shan HM, Shi YX, Quan JM. Identification of green tea catechins as potent inhibitors of the polo-box domain of polo-like kinase 1. ChemMedChem, 2015, 10(1): 158-163. DOI:10.1002/cmdc.201402284 |
| [46] | Chen YY, Zhang J, Li DS, et al. Identification of a novel Polo-like kinase 1 inhibitor that specifically blocks the functions of Polo-Box domain. Oncotarget, 2017, 8(1): 1234-1246. DOI:10.18632/oncotarget.13603 |
| [47] | Lee KS, Grenfell TZ, Yarm FR, et al. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc Natl Acad Sci USA, 1998, 95(16): 9301-9306. DOI:10.1073/pnas.95.16.9301 |
| [48] | Yun SM, Moulaei T, Lim D, et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol, 2009, 16(8): 876-882. DOI:10.1038/nsmb.1628 |
| [49] | Liu F, Park JE, Qian WJ, et al. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem Biol, 2012, 7(5): 805-810. DOI:10.1021/cb200469a |
| [50] | Liu F, Park JE, Qian WJ, et al. Peptoid-Peptide hybrid ligands targeting the polo box domain of polo-like kinase 1. Chembiochem, 2012, 13(9): 1291-1296. DOI:10.1002/cbic.201200206 |
| [51] | Liu F, Park JE, Qian WJ, et al. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat Chem Biol, 2011, 7(9): 595-601. DOI:10.1038/nchembio.614 |
| [52] | ?led? P, Stubbs CJ, Lang S, et al. From crystal packing to molecular recognition: prediction and discovery of a binding site on the surface of polo-like kinase 1. Angew Chem Int Ed Engl, 2011, 50(17): 4003-4006. DOI:10.1002/anie.201008019 |
| [53] | Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell, 2011, 144(5): 646-674. DOI:10.1016/j.cell.2011.02.013 |
| [54] | Ivanov AA, Khuri FR, Fu HA. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci, 2013, 34(7): 393-400. DOI:10.1016/j.tips.2013.04.007 |
| [55] | Ni D, Liu N, Sheng CQ. Allosteric modulators of protein-protein interactions (PPIs)//Zhang J, Nussinov R. Protein Allostery in Drug Discovery. Singapore: Springe, 2019, 1163: 313-334. |
| [56] | Archambault V, Normandin K. Several inhibitors of the Plk1 Polo-Box domain turn out to be non-specific protein alkylators. Cell Cycle, 2017, 16(12): 1220-1224. DOI:10.1080/15384101.2017.1325043 |
| [57] | Abdeldayem A, Raouf YS, Constantinescu SN, et al. Advances in covalent kinase inhibitors. Chem Soc Rev, 2020, 49(9): 2617-2687. DOI:10.1039/C9CS00720B |
| [58] | Mabonga L, Kappo AP. Protein-protein interaction modulators: advances, successes and remaining challenges. Biophys Rev, 2019, 11(4): 559-581. DOI:10.1007/s12551-019-00570-x |
| [59] | Zhang X, Wang YL, Wang JY, et al. Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer (Review). Oncol Rep, 2016, 35(2): 625-638. DOI:10.3892/or.2015.4464 |
| [60] | Murugan RN, Park JE, Kim EH, et al. Plk1-targeted small molecule inhibitors: molecular basis for their potency and specificity. Mol Cells, 2011, 32(3): 209-220. DOI:10.1007/s10059-011-0126-3 |
| [61] | Erak M, Bellmann-Sickert K, Els-Heindl S, et al. Peptide chemistry toolbox-Transforming natural peptides into peptide therapeutics. Bioorg Med Chem, 2018, 26(10): 2759-2765. DOI:10.1016/j.bmc.2018.01.012 |
