 , 王琦2, 刘丁嘉2, 崔敏姝2, 陆强2
, 王琦2, 刘丁嘉2, 崔敏姝2, 陆强2 , 庄柯1, 杨勇平2
, 庄柯1, 杨勇平21. 国电环境保护研究院, 南京 210031;
2. 华北电力大学生物质发电成套设备国家工程实验室, 北京 102206
收稿日期: 2018-06-19; 修回日期: 2018-08-06; 录用日期: 2018-08-06
基金项目: 国家重点基础研究发展计划"973"计划(No.2015CB251501);北京市科技新星计划(No.Z171100001117064);霍英东教育基金会项目(No.161051)
作者简介: 周锦晖(1988-), 男, E-mail:zhoujinhui02@126.com
通讯作者(责任作者): 陆强(1982—),男,教授,从事固体燃料高效热利用及烟气污染物治理研究. E-mail: qianglu@mail.ustc.edu.cn
摘要: 以纳米TiO2为载体,采用等体积浸渍法掺杂过渡金属氧化物ZrO2进行改性,制备了一系列ZrO2/TiO2催化剂,以催化H2O2低温氧化NO脱硝,并采用X射线衍射(XRD)、H2程序升温还原(H2-TPR)、O2程序升温氧化(O2-TPO)、X射线光电子能谱(XPS)及电子顺磁共振(EPR)等表征分析探究了影响H2O2脱硝活性的因素.表征结果表明ZrO2的负载量会影响催化剂中晶格氧的含量,晶格氧相对含量的增加有利于氧化还原反应中的电子传递,这是促进H2O2活化分解的关键.在微观表征的基础上,通过实验研究筛选获得了催化剂的最佳ZrO2负载量,同时对比考察了非催化和纳米TiO2催化作用下的H2O2氧化低温脱硝性能;针对获取的最优催化剂,进一步考察了不同烟气工况对催化剂活性的影响.实验结果表明,ZrO2/TiO2催化剂能有效促进H2O2的活化分解实现低温脱硝,且ZrO2负载量为4%(质量分数)时,催化活性最高;在烟温为160℃、[H2O2]/[NO]物质的量比为2及空速为30000 h-1时,NO转化率最高可达81%.
关键词:H2O2NO氧化ZrO2/TiO2低温脱硝
Experimental study on low temperature H2O2 oxidative denitrification over ZrO2/TiO2 catalyst
ZHOU Jinhui1
, WANG Qi2, LIU Dingjia2, CUI Minshu2, LU Qiang2 , ZHUANG Ke1, YANG Yongping2 1. State Power Environmental Protection Research Institute, Nanjing 210031;
2. National Engineering Laboratory for Biomass Power Generation Equipment, North China Electric Power University, Beijing 102206
Received 19 June 2018; received in revised from 6 August 2018; accepted 6 August 2018
Abstract: A series of ZrO2/TiO2 catalysts were prepared by incipient wetness impregnation method to catalyze the low-temperature oxidation denitrification by H2O2. The characterization analyses of X-ray diffraction (XRD), H2-temperature programmed reduction (H2-TPR), O2-temperature programmed oxidation (O2-TPO), X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) were performed to explore the factors affecting the denitrification activity of H2O2. The characterization results showed ZrO2 loading will affect the content of the lattice oxygen in the catalyst. The increase of lattice oxygen relative content is beneficial to the electron transfer during redox reaction, which is the key to promote the decomposition of H2O2. The optimum ZrO2 loading of the catalyst was obtained by experimental research. And the performance of ZrO2/TiO2 catalyzed H2O2 oxidative denitrification was compared with that of non-catalytic and nano-TiO2. The effect of flue gas conditions on the catalyst activity was further investigated. The results showed that ZrO2/TiO2 catalyst could effectively promote the decomposition of H2O2 to realize low temperature denitrification. The best catalytic performance was obtained at ZrO2 loading of 4%. Under the flue gas temperature of 160℃, [H2O2]/[NO] molar ratio of 2 and space velocity of 30000 h-1, the NO conversion reached as high as 81%.
Keywords: H2O2NO oxidationZrO2/TiO2low temperature denitrification
1 引言(Introduction)锅炉烟气污染物减排一直是我国大气污染物治理的重要内容.目前广泛使用的烟气脱硝技术是中温NH3选择性催化还原技术(NH3-SCR),此技术脱硝效率高且选择性强,但投资运营成本昂贵,对温度工况要求严格且易发生催化剂中毒(姜烨等, 2013; 李春雨, 2015).从简化系统、降低能耗、绿色高效等角度出发,近年来国内外很多****将目光聚集在了H2O2低温氧化技术.H2O2的氧化作用主要靠其自身均裂生成的自由基,但由于O—O键、O—H键的能量较高,H2O2在没有光、热的条件时一般稳定存在或缓慢分解为O2和H2,要大量生成自由基则需要400 ℃以上的高温(Hiroki et al., 2005).目前,H2O2低温氧化技术主要通过使用合适的催化剂促进H2O2低温下生成羟基自由基,进一步氧化NO转化为NO2、N2O3、N2O5等易溶性的高价态氮氧化物,后经碱液吸收,实现高效脱硝(Ding et al., 2014a).其中催化剂可分为两类,一类是催化剂活性组分为中间价态,如基于铁氧化物的类Fenton催化剂(Fe2O3、CeO2、Co2O3、MnO2、CuO等具有还原态的金属氧化物)(Ding et al., 2014a; 王彦斌等, 2013; 冯勇等, 2013);另一类催化剂活性组分为最高价态,如SiO2、ZrO2以及Al2O3等氧化物(Hiroki et al., 2005; Lousada et al., 2010).然而,****研究表明,应用传统的Fe基催化剂进行烟气脱硝时,存在H2O2消耗量过高及催化剂抗硫性能差的问题,显著限制了此技术的经济性和可行性(Ding et al., 2014b; Huang et al., 2015; Ding et al., 2015);而当采用氧化态为最高价态的氧化物做催化剂,催化效率低下(Miller et al., 1994),不足以满足脱硝需求.
因此,如何在维持高效催化的基础上,降低H2O2的消耗量,提高催化剂的抗硫性能,成为了H2O2低温氧化脱硝技术的研究重点.在众多催化剂中,TiO2作为最高价态金属氧化物,表面具有超亲水性,且Ti—O键极性较大,易于表面羟基的形成,同时,其能够以Haber-Weiss机理催化H2O2活化分解,具有良好稳定性及抗硫性能,受到极大的关注(Matthews, 1984; Suh et al., 2000; Linsebigler et al., 1995).Cahill等(1952)和Massey等(1973)的研究表明由于Ti3+发生的是单电子转移,因此,相比传统Fe2+与H2O2之间的反应,Ti3+与H2O2的反应更易进行.Casuscelli等(2008)开展了Ti-MCM-41催化剂催化H2O2进行高级氧化的研究,结果表明Ti的添加可以有效的增强催化剂上的路易斯酸性位点,利于氧化反应进行,高效去除污染物.基于TiO2的良好表现,本文尝试在TiO2的基础上进行掺杂改性,以期获得新型的高效H2O2活化分解脱硝催化剂.
Lousada等(2012)利用密度泛函理论对H2O2在ZrO2、TiO2及Y2O3表面的分解活化焓进行了理论研究,发现ZrO2比TiO2催化所需的反应活化能更低,且ZrO2具有高表面积、优异的抗硫中毒性能.本研究选用ZrO2对TiO2进行掺杂改性,采用等体积浸渍法制备了不同负载量的ZrO2/TiO2催化剂,并结合XRD、TPR、XPS、EPR等微观表征分析催化剂的理化性质.此外,对ZrO2/TiO2催化剂的最佳负载量进行筛选,得到了催化H2O2氧化脱硝的高效催化剂,并与非催化及纳米TiO2催化条件下的H2O2低温氧化脱硝进行活性对比.在此基础上,在模拟烟气条件下考察最佳负载量的ZrO2/TiO2催化剂对不同烟气工况影响,结合催化剂的表征,对实验结果进行分析说明,为发展改性TiO2催化剂在H2O2催化氧化低温脱硝技术中的应用提供数据支撑.
2 实验部分(Experimental)2.1 催化剂的制备以纳米TiO2(江苏汇鸿国际集团中锦控股有限公司)为载体,ZrClO2·8H2O(阿拉丁,纯度99%)为ZrO2的前驱物,采用等体积浸渍法制备所需催化剂.具体制备方法如下:称取一定量的ZrClO2·8H2O并配置成溶液,将溶液加入至纳米TiO2粉末中,搅拌均匀后超声振荡2 h,并在室温下浸渍24 h,在110 ℃下干燥12 h、550 ℃焙烧3 h后获得x%ZrO2/TiO2催化剂,其中x%为ZrO2的质量分数,催化剂记为xZrO2/TiO2.另使用非催化以及纳米TiO2催化作为参照组.
2.2 催化剂的表征采用氮气物理吸附法(ASAP2020型物理吸附仪)对催化剂进行比表面积和孔容、孔径的测定,先将样品分别在90 ℃下进行1 h及200 ℃下进行6 h的抽真空处理;在液氮温度下根据静态法进行材料吸附-脱附等温线的测量;采用BET方程计算催化剂的比表面积,采用BJH方法计算催化剂的孔容、孔径.
采用日本理学公司D/max-ⅢA型全自动X射线衍射仪对催化剂进行XRD分析,辐射源为Cu Ka,测试波长为0.15406 nm,在30 kV/30 mA条件下进行工作,在10°~90°区间内扫描速度为8°·min-1.
采用ChemBET Pulsar TPR/TPD型多功能化学吸附分析仪对催化剂进行程序升温还原(H2-TPR)和程序升温氧化(O2-TPO)分析,对比不同催化剂的氧化还原性能以及储氧能力.测试温度范围为50~900 ℃,升温速率为10 ℃·min-1.
采用Thermo escalab 250Xi型X射线光电子能谱仪对催化剂进行X射线光电子能谱分析(XPS),以单色Al Kα(hv =1 486. 6 eV)为射线源,功率150 W,500 μm束斑,以表面污染碳的C 1s电子结合能(284. 8 eV)进行校准.
采用电子顺磁共振(EPR)光谱仪对反应过程中的自由基中间体进行检测,使用5, 5-二甲基-1-吡咯啉N-氧化物(DMPO, Sigma-Alrich)对自由基进行捕获,测试中设置调制频率为100 kHz,调制振幅为4 G,谐振频率为9.87 GHz,扫描宽度为200 G,微波功率为19.22 mW,中心场为3522 G.
2.3 催化剂活性评价催化剂经研磨、筛分获得粒径为60~80目(0.18~0.25 mm)的粉末催化剂,采用固定床反应器进行脱硝性能评价,实验装置示于图 1.实验中,将催化剂固定于反应器中部,模拟烟气的各路气体流量经由质量流量计控制,并在混气罐内混合均匀后进入固定床反应器.H2O2溶液流量由蠕动泵进行控制,经雾化器进行雾化.尾气吸收系统采用饱和NaOH溶液作为吸收液.反应器进出口处模拟烟气中各气体浓度采用ABB公司MB3000型傅里叶红外烟气分析仪进行分析测试.
图 1(Fig. 1)
 |
| 图 1 H2O2催化氧化脱硝实验台示意图 Fig. 1Schematic diagram of the H2O2-catalyzed oxidative denitrification setup |
实验采用5%的H2O2溶液,由30%的H2O2溶液配制而成.通过H2O2流量的变化来控制烟气中H2O2与NO的物质的量比,记作[H2O2]/[NO]物质的量比.催化剂活性评价指标定义为NO转化率,采用式(1)进行计算.
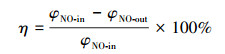 | (1) |
实验所用标准烟气工况如下:温度为160 ℃,NO为500 mg·m-3,SO2为1000 mg·m-3,O2体积分数为3%,N2为平衡气,总流量为3 L·min-1,空速为30000 h-1,[H2O2]/[NO]物质的量为2.
3 结果与讨论(Results and discussion)3.1 催化剂的表征3.1.1 比表面积及孔结构表征表 1列出了xZrO2/TiO2催化剂的比表面积、孔容和平均孔径随ZrO2负载量的变化情况.从表中可以看出,当ZrO2含量从0增加到6%时,催化剂的比表面积和孔容先下降后升高:在ZrO2掺杂量为0~4%时,比表面积逐渐降低,这也许可以归结为部分沉积的ZrO2渗透到载体的孔道中,导致孔道发生部分堵塞;而ZrO2掺杂量继续增加时,比表面积转而变为增加.这种上升的趋势与其他****的对于ZrO2大量掺杂TiO2后的复合金属催化剂比表面积会有效增加的结果相符合(张亚平等, 2016; 2015).
表 1(Table 1)
| 表 1 xZrO2/TiO2催化剂的比表面积和孔结构 Table 1 Specific surface areas and pore structures of xZrO2/TiO2 catalysts | |||||||||||||||||||||||||||||||||||
表 1 xZrO2/TiO2催化剂的比表面积和孔结构 Table 1 Specific surface areas and pore structures of xZrO2/TiO2 catalysts
| |||||||||||||||||||||||||||||||||||
3.1.2 XRD纳米TiO2及ZrO2掺杂的纳米TiO2的XRD谱图如图 2a所示.从图中可以看出,纳米TiO2为锐钛矿型结构,其主要衍射峰的衍射角2θ分别为25.29°、37.80°、48.00°、53.84°和62.75°.xZrO2/TiO2负载型双金属氧化物催化剂的XRD谱图仍以锐钛矿相结构的TiO2衍射峰为主,并没有看到ZrO2的衍射峰,这可能是由于催化剂中ZrO2的含量较低,其有可能为无定形态,在TiO2表面实现了高度分散.通过对锐钛矿型TiO2的最强衍射峰(101)应用Scherrer公式可以估算出催化剂的平均粒径.对于纳米TiO2以及ZrO2掺杂的TiO2样品,经计算获得了催化剂平均晶粒尺寸为3~4 nm.另外,由图 2b可知,随着ZrO2掺杂量的增加,其特征衍射峰稍有变化,衍射峰的2θ角向右发生偏移,以TiO2在25.29°处的最强衍射峰为例,ZrO2掺杂量为0、2%、4%及6%时,其对应的2θ角分别由24.28°偏移至25.31°、25.36°及25.39°,且峰强以及结晶度均有所增大.一方面,这可能是催化剂中有自由缺陷的OH基团发生缩合以形成具有高百分比结晶度的xZrO2/TiO2.另一方面,根据现代晶体学理论,当杂质和基质的离子半径和电负性相近时,形成杂质替位的几率很大,晶相图谱表现为同一方向微弱的移动,晶格间距膨胀或缩小.已知Zr4+离子半径(0.79 ?)接近Ti4+的离子半径(0.74 ?),且Zr4+和Ti4+的电负性(鲍林标度)分别为1.33和1.54,相对比较接近,因此掺杂的Zr4+可以以间隙引入或同质取代的方式进入TiO2晶格中(孙传智, 2011),由于Zr4+的有效离子半径大于Ti4+,造成晶格中氧离子分布的变化,产生电荷补偿的缺陷位点,这些缺陷位点可以捕获电子,能够提高催化剂的氧化还原性能(Hwang et al., 2017).
图 2(Fig. 2)
 |
| 图 2 xZrO2/TiO2催化剂的XRD谱图(a)及在2θ为24°~27°范围内的XRD谱图(b) Fig. 2XRD patterns of xZrO2/TiO2 catalysts (a) and XRD patterns in the range of 2θ at 24°~27° (b) |
3.1.3 H2-TPR和O2-TPO纳米TiO2及ZrO2掺杂的纳米TiO2催化剂的H2-TPR曲线如图 3a所示.由图 3a可知,xZrO2/TiO2系列催化剂均只有一个还原峰.在579 ℃出现的耗氢峰对应着TiO2的还原(Zhu et al., 2004);ZrO2/TiO2系列催化剂相对TiO2还原峰向高温方向移动,且其还原温度随掺杂Zr的质量分数的增加呈先增后减的趋势.这一结果表明TiO2掺杂ZrO2后,其还原活性相较于TiO2有所降低,在ZrO2掺杂量为2%~4%时,催化剂还原活性低于ZrO2掺杂量为5%~6%的催化剂.但通过H2-TPR曲线的还原峰面积可明显看出,4ZrO2/TiO2催化剂具有最大的峰面积,对应着此催化剂的总耗氢量最多,这表明该催化剂储氧能力最强,能够最大程度的增强表面晶格中活性氧的传递和还原过程,促进氧化还原反应进行以及羟基自由基的产生(Mars et al., 1954).
图 3(Fig. 3)
 |
| 图 3 xZrO2/TiO2催化剂的H2-TPR谱图(a)和O2-TPO谱图(b) Fig. 3H2-TPR spectra (a) and O2-TPO spectra (b) of xZrO2/TiO2 catalysts |
纳米TiO2以及ZrO2掺杂的纳米TiO2催化剂的O2-TPO曲线如图 3b所示.由TPO谱图可以看出,在600~900 ℃的TPO过程中,各催化剂均出现了一个耗氧峰,且添加了ZrO2的纳米TiO2催化剂的耗氧峰发生了明显增大.说明ZrO2的掺杂能够明显增强催化剂的氧化还原能力,且随着ZrO2掺杂量的增大,其氧化温度先增大后减小,这结果与TPR的结果相符,说明催化剂的氧化还原性可逆.
3.1.4 XPS图 4a、4b和4c分别给出了xZrO2/TiO2系列催化剂的Ti2p、Zr3d及O1s的XPS谱图.从图 4a中可以见,Ti2p区域可分解为Ti3+(459.50 eV)和Ti4+(459.02 eV和464.83 eV)(Wang et al., 2012; Guillemot et al., 2002).利用峰面积来估算Ti3+和Ti4+的含量,结果示于表 2中.从表中可以看出Zr掺杂会导致Ti3+的增加:当Zr掺杂量由0增加至4%时,Ti3+含量由9.94%增加至10.99%,但随着ZrO2含量继续增加时,Ti3+的浓度逐渐降低.由前人研究发现,Ti3+对于产生羟基自由基的催化反应具有促进作用.除此之外,Ti2p3/2峰位置随着ZrO2掺杂量的增加向低结合能方向移动,这可能是由于Zr4+将电子传递给Ti4+造成的(Li et al., 2007).
图 4(Fig. 4)
 |
| 图 4 xZrO2/TiO2催化剂的Ti2p(a)、Zr3d(b)和O1s(c)XPS谱图 Fig. 4Ti2p (a), Zr3d (b) and O1s (c) XPS spectra of xZrO2/TiO2 catalysts |
表 2(Table 2)
| 表 2 xZrO2/TiO2催化剂中Ti(Ⅲ)和Ti(Ⅳ)以及OⅠ和OⅡ的相对含量 Table 2 Relative contents of Ti(Ⅲ), Ti(Ⅳ) and OⅠ, OⅡ in the xZrO2/TiO2 catalysts | ||||||||||||||||||||||||||||||||||||||||
表 2 xZrO2/TiO2催化剂中Ti(Ⅲ)和Ti(Ⅳ)以及OⅠ和OⅡ的相对含量 Table 2 Relative contents of Ti(Ⅲ), Ti(Ⅳ) and OⅠ, OⅡ in the xZrO2/TiO2 catalysts
| ||||||||||||||||||||||||||||||||||||||||
如图 4b所示,对于xZrO2/TiO2系列催化剂,Zr3d5/2的结合能在182~183 eV范围内,随着ZrO2掺杂量的增多,结合能分别为182.60、182.71和182.58 eV,可见结合能随Zr掺杂量增加先向高结合能方向偏移,随后恢复到低结合能.这表明Zr4+周围电子密度先降低后增加.据此可以推断,4ZrO2/TiO2催化剂中可能形成了Ti—O—Zr键,并且在Ti—O—Zr体系中,Ti4+离子吸引了附近氧原子的电子,从而造成Zr4+周围电子缺失以及Ti4+周围电子富集,从而在晶格中形成了大量的缺陷位点(Pérez-Hernández et al. 2008).这与XRD的分析结果相一致.
图 4c中O1s有两个信号峰,530 eV附近的信号峰归属于晶格氧OⅠ,而532 eV处的信号峰归属于化学吸附氧OⅡ(Min et al., 2007).跟据Mars-Van Krevelen理论,氧化还原反应催化剂多为过渡金属氧化物MOn,而氧化剂多为O2,氧化物中晶格氧与反应物反应,形成氧空位MOn-1,O2再吸附在氧化物上重新形成MOn,从而完成催化循环(Mars et al., 1954).部分xZrO2/TiO2催化剂中晶格氧OⅠ以及化学吸附氧OⅡ所占百分比如表 2所示.4ZrO2/TiO2催化剂具有最高的表面晶格氧占比,从而会在催化剂表面形成最多的氧空位,促进反应中的电子转移,形成最多的羟基自由基,且能促使自由基吸附,从而延长自由基半衰期(Yang et al., 2015).
3.1.5 EPR图 5给出了纯H2O2体系、TiO2催化H2O2体系以及4ZrO2/TiO2催化H2O2体系中测定的·OH自由基的EPR谱图.图中测量出的实际谱线为典型的四重精细结构,各峰强度比为1:2:2:1(aN=aH=14.9 G),对应的是典型的DMPO-·OH加合物的波普特征信号峰(叶苗苗等, 2008).据此可以证实H2O2的活化分解产物主要是·OH,且·OH是促进NO氧化的主要中间物质.由图可知,在无催化剂、TiO2催化作用,以及4ZrO2/TiO2催化作用下的H2O2体系中,DMPO-·OH信号峰峰强发生显著提高,说明了这3种不同体系中·OH的产量逐渐增加.
图 5(Fig. 5)
 |
| 图 5 不同反应体系的EPR谱图 Fig. 5EPR spectra in different reaction systems |
3.2 催化剂活性评价3.2.1 催化剂组成对脱硝性能的影响图 6给出了不同负载量的ZrO2/TiO2催化剂在160、200、240 ℃条件下的NO转化率.由图 6可知,随着ZrO2负载量增加,NO转化率呈先增后减的趋势;当ZrO2负载量为4%时,NO转化率达到峰值.其中,温度为160 ℃时,4ZrO2/TiO2催化作用下NO转化率可达80%.当温度升高时,4ZrO2/TiO2的催化效率发生降低,当温度为200 ℃和240 ℃时,NO转化率分别降为77%和70%.
图 6(Fig. 6)
 |
| 图 6 xZrO2/TiO2催化剂中ZrO2负载量对NO转化率的影响 Fig. 6Effect of ZrO2 loading in the xZrO2/TiO2 catalyst on NO conversion |
图 7给出了120~280 ℃条件下,非催化、TiO2催化及4ZrO2/TiO2催化作用下,H2O2低温氧化脱硝的NO转化率.在高温区间内,非催化作用下H2O2的活化分解是****们早期研究的主题.然而,从图中可以看出,在低温区间内,NO转化率最高仅26%,且随温度上升而效率提高,表明H2O2的均裂活化能高,需要通过高温或其他催化手段来促进其活化分解.进一步以纳米TiO2及掺杂改性后的4ZrO2/TiO2为催化剂,促进低温下H2O2氧化脱硝.由结果可知,引入TiO2催化剂可显著提升H2O2的氧化脱硝能力,但NO转化率随温度变化波动较大,难以满足工业烟气脱硝的需求;改性后的ZrO2/TiO2催化剂,在TiO2催化的基础上再次提高了脱硝效率,且在整个低温段(120~280 ℃)的NO转化率都保持在65%以上,最高可达81%.
图 7(Fig. 7)
 |
| 图 7 无催化剂、TiO2作用下及4ZrO2/TiO2作用下的NO转化率 Fig. 7NO conversion under non-catalysis as well as TiO2 and 4ZrO2/TiO2-catalyzed conditions |
3.2.2 催化剂抗硫性能评价烟气中一定浓度的SO2能在不同的温度下对催化剂脱硝反应起到促进或抑制作用,图 8a给出了初始SO2浓度分别为600、800、1000和1200 mg·m-3时对4ZrO2/TiO2催化剂脱硝性能的影响.当SO2浓度为1000 mg·m-3及以下时,SO2的存在对催化剂脱硝效率几乎没有影响;而SO2浓度为1200 mg·m-3时,SO2造成催化剂活性有一定幅度的下降,NO转化率从初始的81%下降至77%.这可能是由于在低温条件下,SO2浓度过高时,SO2会与NO产生竞争吸附作用.NO转化率下降幅度小,表明ZrO2改性后的催化剂在低温区域对较高浓度SO2烟气条件的适应性也有所提高.
图 8(Fig. 8)
 |
| 图 8 SO2入口浓度对NO转化率的影响(a)及4ZrO2/TiO2催化剂抗硫稳定性实验(b) Fig. 8Effect of SO2 inlet concentration (a) on NO conversion and experimental study on the stability of 4ZrO2/TiO2 catalyst for sulfur resistance (b) |
图 8b给出了4ZrO2/TiO2催化剂在初始SO2浓度为500 mg·m-3的条件下,时长为30 h的抗硫稳定性测试的实验结果.由图可以看出,4ZrO2/TiO2催化剂经过30 h反应后,脱硝效率依然能够保持稳定.在催化剂工作的30 h内,NO氧化率仅存在误差范围内的波动,NO氧化率的最高值及最低值分别为81%和77%.由上述结果可知,4ZrO2/TiO2催化剂具有优异的抗SO2中毒稳定性能.
3.2.3 烟气条件对催化剂脱硝性能的影响鉴于4ZrO2/TiO2催化剂具有优异的催化性能,为深入了解催化剂对不同烟气工况的适应性,进一步研究了[H2O2]/[NO]物质的量比、NO入口浓度、氧量和空速等因素对催化剂性能的影响.
图 9a给出了[H2O2]/[NO]物质的量比对催化剂性能的影响.通过控制H2O2流量可以实现[H2O2]/[NO]物质的量比的控制.由图可知,在[H2O2]/[NO]物质的量比为0.5~3范围内时,NO转化率随[H2O2]/[NO]物质的量比增大呈现出同步提升,但随着[H2O2]/[NO]物质的量比继续增大,NO转化率反而降低.在[H2O2]/[NO]物质的量比为3时,NO转化率最高可达85%.这种现象产生的原因可能是·OH氧化NO的反应主要发生在催化剂表面,而[H2O2]/[NO]物质的量比增加过大,会导致催化剂表面的浸湿,从而溶解了部分羟基自由基,导致羟基自由基无法与NO接触,进而影响NO氧化反应的进行.由此可知,选择合适的[H2O2]/[NO]物质的量比对于H2O2低温脱硝技术的经济性具有重要影响.
图 9(Fig. 9)
 |
| 图 9 [H2O2]/[NO]物质的量比(a)、NO入口浓度(b)、O2入口含量(c)及空速(d)对NO转化率的影响 Fig. 9Effect of [H2O2]/[NO] molar ratio (a), NO inlet concentration (b), O2 inlet concentration (c) and space velocity on NO conversion |
图 9b给出了不同初始NO浓度对4ZrO2/TiO2催化剂催化活性的影响.由图可知,NO浓度为400、500、600和700 mg·m-3时,催化剂脱硝效率分别为82%、80%、75%和72%,随着NO浓度增加,催化剂脱硝效率略有下降.鉴于不同锅炉排放的烟气中NO浓度有所差异,可通过适当调整[H2O2]/[NO]物质的量比来抑制NO入口浓度对催化剂性能的负面影响.
图 9c给出了不同入口氧量对4ZrO2/TiO2催化剂脱硝效率的影响.由图可知,随着烟气中氧量含量由0增加到3%(体积分数),NO氧化率有一个激增,由74%增加到80%,表明烟气中氧气组分对催化活性有较大影响.此影响可能体现在催化剂的化学吸附氧量上,当烟气中含有氧气时,催化剂表面可吸附一定量的氧气,从而促进氧化还原反应进行以及羟基自由基的形成.在烟气中氧气的体积含量由3%增加到9%时,NO氧化率基本无变化,可见烟气中过多的氧气不会增加催化剂表面的化学吸附氧量.也证实了该催化剂对不同氧含量具有良好的适应性.
图 9d给出了不同空速对4ZrO2/TiO2催化剂催化活性的影响.由图可知,随着空速的增加催化剂的脱硝效率稍有降低,当空速从10000 h-1提高到70000 h-1,催化剂脱硝效率从81%下降至79%.结果表明,4ZrO2/TiO2催化剂脱硝效率对空速的敏感程度较低,适应性很强.
4 结论(Conclusions)1) 改性后的4ZrO2/TiO2催化剂显著提升了NO转化率,并具有优良的抗硫中毒能力,连续测试30 h,脱硝效率始终保持在81%左右;同时该催化剂对温度、[H2O2]/[NO]物质的量比、NO浓度、SO2浓度、O2浓度、空速等烟气条件均具有良好的适应性.
2) 氧空位或缺陷位点数量是ZrO2/TiO2负载型催化剂活性的关键影响因素,氧空位或缺陷位点是H2O2活化分解的主要活性中心,可以吸附·OH并延长自由基半衰期.
3) 催化剂储氧量越高、表面晶格氧含量占比越大,ZrO2/TiO2催化剂活性越高.
参考文献
| Cahill A E, Taube H. 1952. The use of heavy oxygen in the study of reactions of hydrogen peroxide[J]. Journal of the American Chemical Society, 74(9): 2312–2318.DOI:10.1021/ja01129a042 |
| Casuscelli S G, Eimer G A, Canepa A, et al. 2008. Ti-MCM-41 as catalyst for α-pinene oxidation:Study of the effect of Ti content and H2O2 addition on activity and selectivity[J]. Catalysis Today, 133(11): 678–683. |
| Ding J, Zhong Q, Zhang S L. 2014a. Simultaneous removal of NOx and SO2 with H2O2 over Fe based catalysts at low temperature[J]. RSC advances, 4(11): 5394–5398.DOI:10.1039/c3ra46418k |
| Ding J, Zhong Q, Zhang S L, et al. 2014b. Simultaneous removal of NOx and SO2 from coal-fired fue gas by catalytic oxidation-removal process with H2O2[J]. Chemical Engineering Journal, 243(5): 176–182. |
| Ding J, Zhong Q, Zhang S L, et al. 2015. Size-and shape-controlled synthesis and catalytic performance of iron-aluminum mixed oxide nanoparticles for NOx and SO2 removal with hydrogen peroxide[J]. Journal of Hazardous Materials, 283: 633–642.DOI:10.1016/j.jhazmat.2014.10.010 |
| 冯勇, 吴德礼, 马鲁铭. 2013. 铁氧化物催化类Fenton反应[J]. 化学进展, 2013, 25(7): 1219–1228. |
| Guillemot F, Porte M C, Labrugère C, et al. 2002. Ti4+ to Ti3+ conversion of TiO2 uppermost layer by low-temperature vacuum annealing:interest for titanium biomedical applications[J]. Journal of colloid and interface Science, 255(1): 75–78.DOI:10.1006/jcis.2002.8623 |
| Hiroki A, Laverne J A. 2005. Decomposition of hydrogen peroxide at water-ceramic oxide interfaces[J]. Journal of Physical Chemistry B, 109(8): 3364–3370.DOI:10.1021/jp046405d |
| Huang X M, Ding J, Zhong G Q. 2015. Catalutic decomposition of H2O2 over Fe-based catalysts for simultaneous removal of NOx and SO2[J]. Applied Surface Science, 326: 66–72.DOI:10.1016/j.apsusc.2014.11.088 |
| Hwang J, Rao R R, Giordano L, et al. 2017. Perovskites in catalysis and electrocatalysis[J]. Science, 358(6364): 751–756.DOI:10.1126/science.aam7092 |
| 姜烨, 高翔, 吴卫红, 等. 2013. 选择性催化还原脱硝催化剂失活研究综述[J]. 中国电机工程学报, 2013, 33(14): 18–31. |
| 李春雨. 2015. 我国火电厂SCR烟气脱硝技术研究及应用综述[J]. 能源环境保护, 2015, 29(5): 8–12.DOI:10.3969/j.issn.1006-8759.2015.05.002 |
| Li J, Zeng H C. 2007. Hollowing Sn-doped TiO2 nanospheres via ostwald ripening[J]. Journal of the American Chemical Society, 129(51): 15839–15847.DOI:10.1021/ja073521w |
| Linsebigler A L, Lu G Q, Yates J T. 1995. Photocatalysis on TiO2 surfaces:Principles, mechanisms, and selected results[J]. Chemical Reviews, 95(3): 735–758.DOI:10.1021/cr00035a013 |
| Lousada C M, Johansson A J, Brinck T, et al. 2012. Mechanism of H2O2 decomposition on transition metal oxide surfaces[J]. Journal of Physical Chemistry C, 116(17): 9533–9543.DOI:10.1021/jp300255h |
| Lousada C M, Jonsson M. 2010. Kinetics, mechanism, and activation energy of H2O2 decomposition on the surface of ZrO2[J]. Journal of Physical Chemistry C, 114(25): 11202–11208.DOI:10.1021/jp1028933 |
| Mars P, Van Krevelen D W. 1954. Oxidations carried out by means of vanadium oxide catalysts[J]. Chemical Engineering Science, 3: 41–59.DOI:10.1016/S0009-2509(54)80005-4 |
| Massey V, Palmer G, Ballou D P, et al. 1973. Oxidases and related redox systems[J]. by King T E, Mason H S, Morrison M, Univ. Park Press, Baltimore, 1: 25. |
| Matthews R W. 1984. Hydroxylation reactions induced by near UV photolysis of aqueous titanium dioxide suspensions[J]. Journal of the Chemical Society Faraday Transactions, 80(2): 457–471.DOI:10.1039/f19848000457 |
| Miller J B, Rankin S E, Ko E I. 1994. Strategies in controlling the homogeneity of zirconia-silica aerogels:Effect of preparation on textural and catalytic properties[J]. Journal of Catalysis, 148(2): 673–682.DOI:10.1006/jcat.1994.1254 |
| Min K, Park E D, Ji M K, et al. 2007. Manganese oxide catalysts for NOx reduction with NH3 at low temperatures[J]. Applied Catalysis A:General, 327(2): 261–269.DOI:10.1016/j.apcata.2007.05.024 |
| Pérez-Hernández R, Mendoza-Anaya D, Fernández M E, et al. 2008. Synthesis of mixed ZrO2-TiO2, oxides by sol-gel:Microstructural characterization and infrared spectroscopy studies of NOx[J]. Journal of Molecular Catalysis A Chemical, 281(1): 200–206. |
| Suh M, Bagus P S, Pak S, et al. 2000. Reactions of hydroxyl radicals on titania, silica, alumina, and gold surfaces[J]. Journal of Physical Chemistry B, 104(12): 2736–2742.DOI:10.1021/jp993653e |
| 孙传智. 2011. TiO2基催化剂的制备、表征及其在环境催化中应用的基础研究[D].南京: 南京大学http://cdmd.cnki.com.cn/Article/CDMD-10284-1016004517.htm |
| Wang W, Lu C H, Ni Y R, et al. 2012. Enhanced visible-light photoactivity of {001} facets dominated TiO2 nanosheets with even distributed bulk oxygen vacancy and Ti3+[J]. Catalysis Communications, 22: 19–23.DOI:10.1016/j.catcom.2012.02.011 |
| 王彦斌, 赵红颖, 赵国华, 等. 2013. 基于铁化合物的异相Fenton催化氧化技术[J]. 化学进展, 2013, 25(8): 1246–1259. |
| Yang M, Jonsson M. 2015. Surface reactivity of hydroxyl radicals formed upon catalytic decomposition of H2O2 on ZrO2[J]. Journal of Molecular Catalysis A:Chemical, 400: 49–55.DOI:10.1016/j.molcata.2015.02.002 |
| 叶苗苗, 陈忠林, 沈吉敏, 等. 2008. 臭氧提高纳米TiO2光催化活性的ESR分析[J]. 影像科学与光化学, 2008, 26(6): 460–467. |
| 张亚平, 郭婉秋, 王龙飞, 等. 2015. V2O5-CeO2/TiO2-ZrO2催化剂表征及NH3还原NOx性能[J]. 催化学报, 2015, 36(10): 1701–1710. |
| 张亚平, 王龙飞, 李娟, 等. 2016. V2O5-WO3/TiO2-ZrO2脱硝催化剂中ZrO2和WO3的促进作用:催化性能、形态及反应机理[J]. 催化学报, 2016, 37(11): 1918–1930. |
| Zhu H, Qin Z, Shan W, et al. 2004. Pd/CeO2-TiO2 catalyst for CO oxidation at low temperature:a TPR study with H2 and CO as reducing agents[J]. Journal of Catalysis, 225(2): 267–277.DOI:10.1016/j.jcat.2004.04.006 |
