为此,动物进化适应与濒危物种保护学科组利用最大熵模型(MaxEnt)对高原鼢鼠和甘肃鼢鼠的适宜栖息地空间分布及重叠分布区进行了模拟分析,在控制大环境相似的前提下,在同域分布区进行了采样,并对两物种胃肠道不同部位以及洞道土壤微生物进行16S rRNA V3-V4区测序,利用快速准确的微生物来源分析(FEAST)方法对两种鼢鼠不同胃肠道部位微生物进行了溯源分析。结果指出,两种鼢鼠胃肠道不同部位微生物组成均存在部位特异性,且盲肠的微生物多样性高于直肠、小肠和胃;两种鼢鼠整个胃肠道及不同部位的微生物的组成均存在显著的种间差异,这种差异可能由宿主遗传背景以及环境因素差异造成。FEAST分析表明,两种鼢鼠胃肠道各部位微生物受洞道土壤影响均很小,同时两种鼢鼠直肠对胃微生物贡献率均在84%左右,推测两种鼢鼠均可能存在食粪行为。基于α多样性、PCoA以及溯源分析结果,认为用粪便样品代替盲肠内容物来进行鼢鼠肠道微生物多样性相关方面的研究是可行的。本研究结果为进一步研究两种鼢鼠不同胃肠道部位微生物群落的功能差异提供了重要的基础信息。
研究以Microbial Biogeography along the Gastrointestinal Tract Segments of Sympatric Subterranean Rodents (Eospalax baileyi and Eospalax cansus)为题近期发表于动物学科专业期刊Animals(二区)。博士研究生刘道鑫为论文的第一作者,张同作研究员为通讯作者。该研究得到了中国科学院-青海省人民政府三江源国家公园联合研究专项和青海省应用基础研究计划等项目资助。学科组依托中科院高原生物适应与进化重点实验室和青海省动物生态基因组学重点实验室。
论文链接:https://doi.org/10.3390/ani11113297
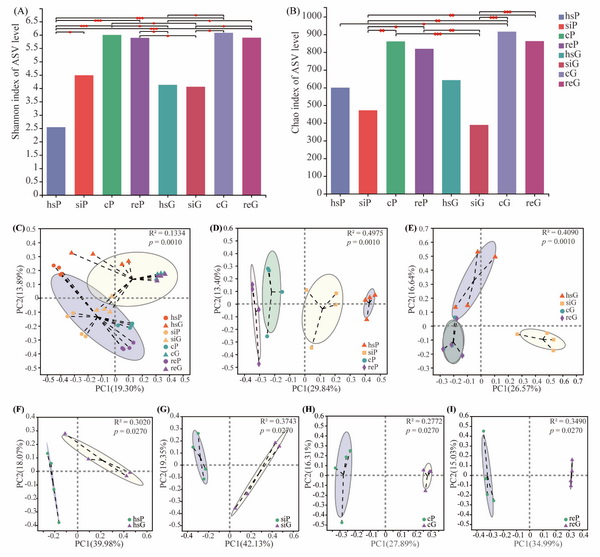
图1同域分布高原鼢鼠和甘肃鼢鼠胃肠道不同部位α多样性差异分析(A-B)和PCoA聚类分析(C-I)
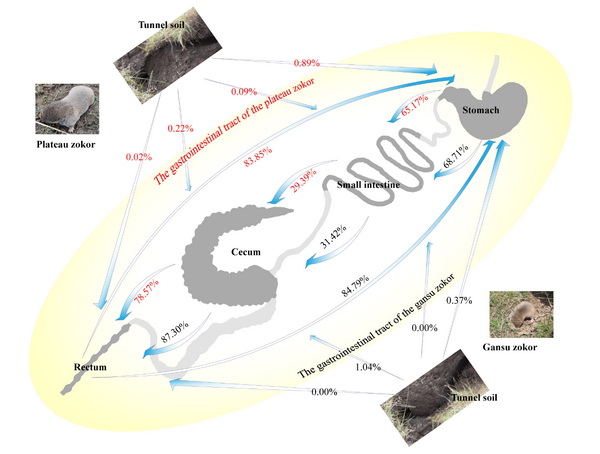
图2同域分布高原鼢鼠和甘肃鼢鼠胃肠道微生物FEAST溯源分析
责任编辑:李暄妍
