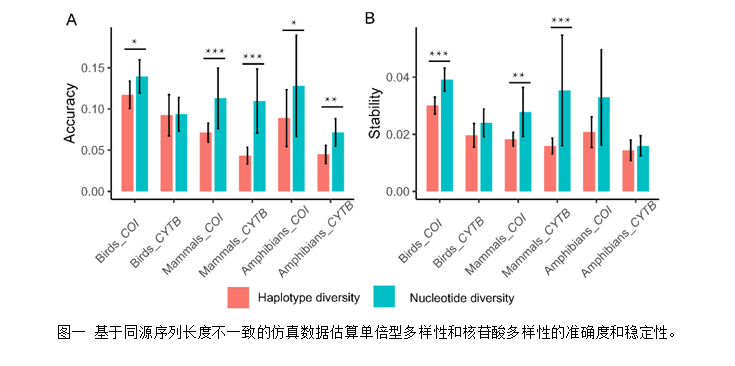
中国科学院动物研究所鸟类学研究组联合哥本哈根大学,提出单倍型多样性计算新方法(公式2)。该方法的优点在于:能够使用同源序列长度不一致的数据对单倍型多样性进行评估;相较于目前宏观遗传多样性格局研究所使用的核苷酸多样性而言,在实际应用中,单倍型多样性在处理不同长度序列上具有较高的精确度和稳定性(图1,精确度以相对误差的均值估算),因此,单倍型多样性更适宜处理序列长度不一致的遗传数据。基于此方法,研究团队进一步探讨了陆生脊椎动物单倍型多样性的分布格局,揭示了不同动物类群南北半球纬度梯度格局的差异性(图2)。在大数据分析时代,本研究提出了基于公共数据库遗传数据评估遗传多样性格局的新方法,为精确评估单倍型多样性以及从多指标评估和理解遗传多样性的格局及其驱动机制提供了可能。

该研究成果以“An approach for estimating haplotype diversity from the sequences with unequal lengths”为题于2021年5月20日在线发表于《Methods in Ecology and Evolution》。中国科学院动物研究所博士生范平为第一作者,中国科学院动物研究所雷富民研究员及宋刚副研究员为通讯作者,哥本哈根大学Fjelds?, Jon教授参与了本研究。该研究得到了中国科学院战略性先导科技专项(A类)、国家自然科学基金、国家留学基金创新型人才国际合作培养等项目的资助。
论文链接:An approach for estimating haplotype diversity from the sequences with unequal lengths
该方法的使用脚本存储于PingFan6/estimating-haplotype-diversity: Estimating haplotype diversity,相关使用说明详见文章支持信息。

责任编辑:张婧睿
