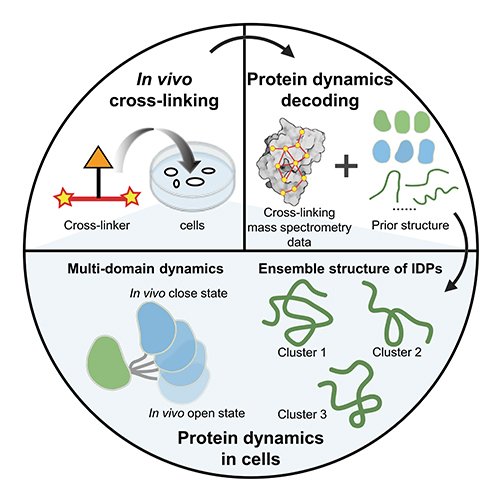
活细胞内蛋白质的原位动态结构对于揭示其生物学功能至关重要。随着深度学习算法助力蛋白质结构预测的发展迭代,AlphaFold2实现了对蛋白质结构的全面预测,然而该方法对柔性区域的结构预测仍面临挑战。近年来,in vivo XL-MS以高通量、高灵敏,且对蛋白质纯度要求低等优势,在解析活细胞内蛋白质的原位动态结构方面展示出重要潜力。张丽华团队一直致力于in vivo XL-MS新技术研究,实现了蛋白质原位构象和相互作用的规模化解析(Anal. Chem.,2020;Anal. Chem.,2022;Anal. Chem.,2022;Anal. Chem.,2022;Anal. Chem.,2023;Angew. Chem. Int. Ed.,2023;Nat. Commun.,2023)。
本工作中,针对多结构域蛋白质,研究团队提出了将结构域作为整体,利用结构域间的XL-MS数据对细胞内蛋白质动态结构建模,实现了三种多结构域蛋白质——钙调蛋白、hnRNP A1和hnRNP D0在细胞内的动态结构表征。此外,针对IDP,研究团队提出了两种互补的结构表征策略:一是将XL-MS信息直接转换为距离约束用于IDP的结构计算,二是首先使用全原子分子动力学模拟进行无偏采样,然后基于XL-MS数据对采样结构进行评估和筛选。利用这两种策略,研究团队解码了高迁移率组蛋白HMG-I/Y和HMG-17在细胞内的动态系综构象。
上述成果以“Decoding Protein Dynamics in Cells Using Chemical Cross-Linking and Hierarchical Analysis”为题,于近日发表在《德国应用化学》(Angewandte Chemie International Edition)。该工作的第一作者是1810组博士研究生张蓓蓉。该工作得到了国家重点研发计划、国家自然科学基金、中国科学院青促会等项目的资助。(文/图 张蓓蓉、赵群、龚洲)
文章链接:https://doi.org/10.1002/anie.202301345
