近日,中国科学院北京基因组研究所(国家生物信息中心)张治华研究组以“DeNOPA: decoding nucleosome positions sensitively with sparse ATAC-seq data”为题在国际生物信息学领域核心期刊Briefings in Bioinformatics上报告了一种基于低测序深度ATAC-seq文库的核小体排布和染色质开放性检测技——deNOPA。不同于现有算法根据测序片段长度,将ATAC-seq文库拆分为分别用于染色质开放性评估和核小体排布检测的子文库的思路,该研究发现,来自不同长度测序片段的Tn5酶切位点,在基因组上分布相似。于是,利用这一相似性,研究人员开发了deNOPA。
该方法创新性的将核小体检测任务的核心问题,由寻找核小体中心转换为寻找核小体连接区域,从而使所有测序片段均在核小体检测任务中得到应用。通过对一系列来自不同物种、不同测序深度ATAC-seq文库,包括单细胞ATAC-seq文库的性能测试。该算法的核小体检测灵敏度相比现有算法大幅度提升,而代价仅是核小体定位准确性的可接受损失。基于该算法给出的核小体位置进行的单细胞分群研究也获得了比现有策略更高的分群精度。
最后,该研究通过对热刺激状态和正常状态K562细胞核小体排布和染色质状态的测定和对比,描述了哺乳动物细胞热刺激反应中染色质状态和核小体排布的变化。不同于酵母,K562细胞热刺激反应中转录起始位点附近核小体缺失区域的位置维持稳定,核小体占位率变化与所在元件活性是否受热刺激影响有关,而与受影响的方向无关。
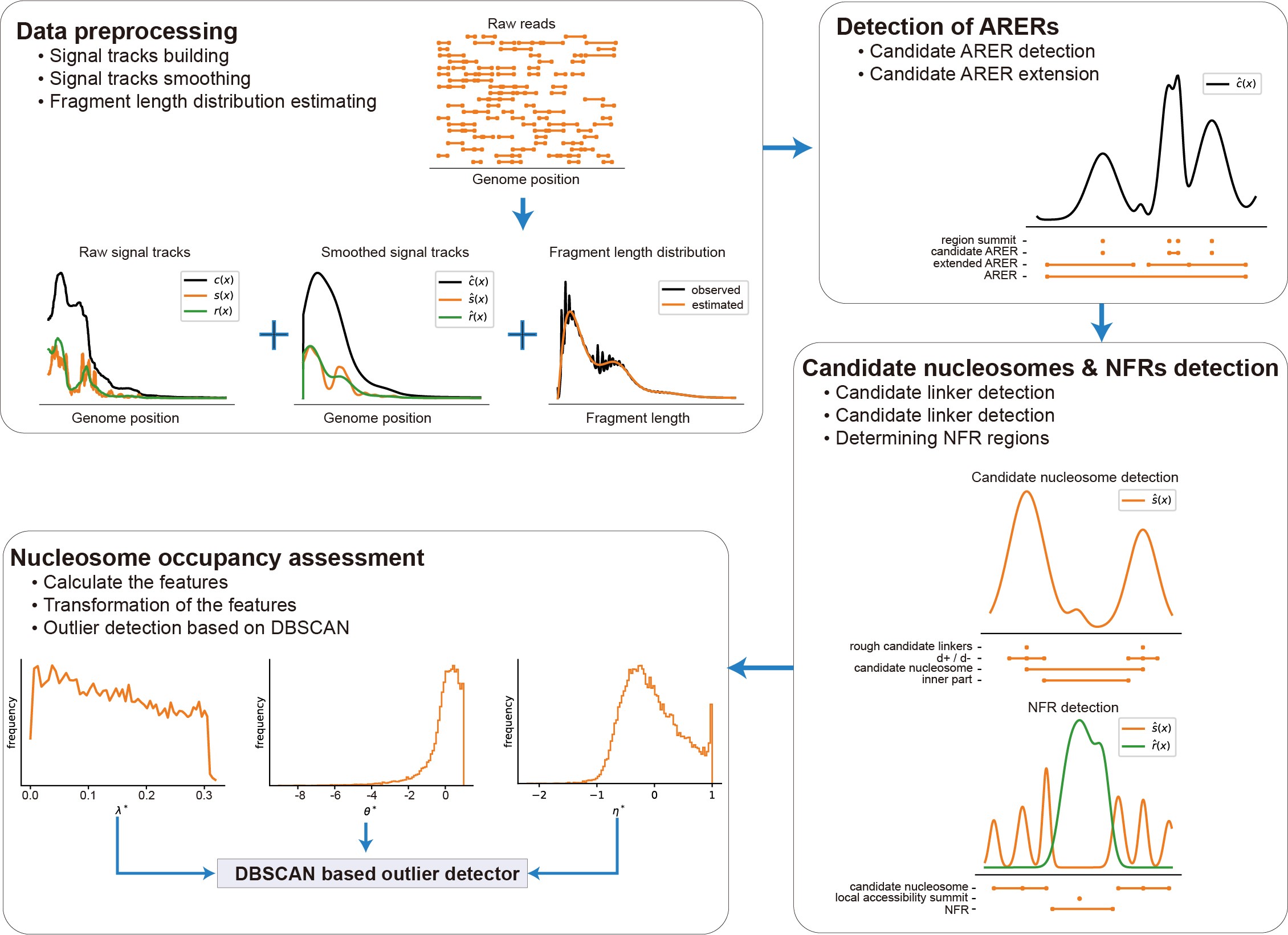
deNOPA算法的处理流程
论文链接
附件下载:
